Análisis conformacional de sulfuros orgánicos que contienen el grupo difenílfosfinoilo mediante sus propiedades atómicas integradas.
Julio
Cárdenas
Universidad La Salle México
Gabriel
Cuevas
Escuela
de
Ciencias
Químicas,Instituto de Química, UNAM.
http://www.chemistry.mcmaster.ca/faculty/bader/aim/
http://www.iquimica.unam.mx/gcuevas.html
RESUMEN
En 1982, el Dr. Eusebio Juaristi y colaboradores descubrieron que el 2-difenilfosfinoil-1,3-ditiano presenta una fuerte preferencia por la conformación
axial. Sin embargo, aún existe controversia sobre cuál es el origen de dicha preferencia conformacional. Se han realizado estudios teóricos y
experimentales que apoyan la existencia de una interacción electrostática del tipo C-H--O-P como el origen de este comportamiento. Con el fin de
contribuir al desarrollo de un modelo general que explique el origen de la preferencia conformacional del 2-difenilfosfinoil-1,3-ditiano se presenta
este trabajo.
Se realizó el estudio de las propiedades atómicas en el marco de la Teoría de Átomos en Moléculas de los confórmeros relevantes del
2-dimetilfosfinoil-1,3-ditiano y el dimetilfosfinoil(metansulfanil) metano, el cual permite conocer la evolución energética de los núcleos con los
cambios conformacionales
Los resultados aquí presentados permiten concluir que la conformación observada experimentalmente es resultado de una competencia entre la
estabilización del sustituyente y de la cadena / anillo
Palabras
Clave:
Cálculos Ab initio, heterociclos, análisis conformacional, Propiedades atómicas • interacciones electrostáticas.
ABSTRACT
In 1982 Juaristi and co-workers reported the pre-dominance of the axial conformation of 2- diphenylphosphinoyl-1,3-dithiane. Although, there is still
controversy about the origin of this preference. Some studies have demostared that electrostatic interactions of type C-H--O-P are the origin.
In order to contribute in the development of a general model that explains the conformational behavior of 2-diphenylphosphinoyl-1,3-dithiane, this
study was performed.
The atomic properties were evaluated within the frame of the Topological Theory of Atoms in Molecules, this study allows to observe how the energy of
every nuclei evolves with conformational changes.
We found that the oberved conformation is result of a competitive stabilization of chain/ring versus substituents
Keywords:
Ab initio calculations, heterocycles, conformation analysis, Atomic properties, electrostatic interactions
EFECTO ANOMÉRICO S-C-P
En 1982 el Dr. Eusebio Juaristi y colaboradores1 descubrieron que el 2-difenilfosfinoil-1,3-ditiano (2-ax) presenta una alta
preferencia por la conformación axial. La diferencia en los desplazamientos químicos de los protones axiales (3.7 ppm) y ecuatoriales (2.8 ppm) de las
posiciones 4,6 es muy alta (1.2 ppm). Juaristi sugirió que dichas observaciones podrían ser debido a un efecto desprotector del grupo fosfinoilo sobre
los protones syn-diaxiales de las posiciones C(4,6), al preferir dicho grupo la conformación axial.
Figura
1.
Preferencia conformacional del 2-difenilfosfinoil-1,3-ditiano
-
 O
O
+
 P PH2 -
P PH2 -

 O
O
 S S
S S
2-ax
DG= 1.0 Kcal/mol
S
S P+Ph
2-eq
La resolución de la estructura cristalina de dicho compuesto por difracción de rayos X fue una prueba irrefutable de que el razonamiento utilizado por
el Dr. Juaristi es el correcto. Una vez resuelta la controversia sobre la preferencia conformacional del 2-difenilfosfinoil-1,3-ditiano, el principal
problema consistía en la explicación de este comportamiento anómalo. Se había afirmado con anterioridad que no era posible la existencia del efecto
anomérico en elementos del segundo periodo de la tabal periódica (Schleyer et al)2 debido a la baja capacidad donadora p del
azufre. Sin embargo, el efecto anomérico en este segmento existe y es, además, uno de los más fuertes reportados hasta ahora (2.64Kcal/mol).3-4 El
efecto anomérico en el segmento S-C-P(O) ha sido explicado por una interacción nS Æ s*C-P, que implica la deslocalización del par de electrones no
compartidos del heteroátomo endocíclico hacia el enlace polar antiperiplanar adyacente.4 De acuerdo con este modelo, debería existir una elongación del
enlace C-P, y un acortamiento del enlace C-S, con respecto al confórmero ecuatorial. Sin embargo, la
determinación de los datos estructurales del 4,6-dimetil-2-difenilfosfinoil-1,3-ditiano, modelo anancomérico de ( 2-ax), y la posterior comparación entre estos dos sistemas, arrojó resultados contrarios a los esperados.5 Por lo que este modelo no
es adecuado para explicar el origen del
efecto anomérico en este sistema en particular.
Tal y como lo corroboran los datos obtenidos por Rayos X, átomo de oxígeno el grupo difenilfosfinoil en (2-ax) se ubica cerca de los átomos de
hidrógeno de los metilenos en posición C(4,6). Por lo que podría existir una fuerte interacción electrostática que estabilice a 2-ax.
Mikolajczyk y colaboradores obtuvieron pruebas experimentales a favor de este argumento,6 al observar que la preferencia por el confórmero 2-ax
disminuye considerablemente en ácido trifluoroacético. Este solvente podría ser capaz de transferir un protón al
oxígeno del grupo fosfinoilo, con lo que se cancelaría cualquier interacción electrostática atractiva entre éste y los
protones syn-axiales en posición C(4,6) de 2-ax. Por otro lado, el 2-difenilfosfinoil-1,3,5-trititiano presenta aún mayor preferencia7 por el
confórmero axial respecto a 2, hecho que es debido al incremento de la acidez de los protones syn-axiales en C(4,6) al sustituirse un
metileno del 2-
difenilfosfinoil-1,3-ditiano por un átomo de azufre, lo que favorece la existencia de interacciones electrostáticas atractivas mayores. Se han
realizado diversos estudios teóricos8 y experimentales9 en derivados de 2-ax donde no es posible la donación de electrones p del
azufre, por lo que el efecto anomérico pretende ser explicado por medio de interacciones electrostáticas como las anteriormente descritas. Tal es el
caso del difenilfosfinoil(metansulfanil)metano10.
El objetivo del presente es dar evidencia de la aplicación general del modelo de interacciones electrostáticas del tipo C-H --- O-P como el origen del
efecto anomérico en el 2- difenilfosfinoil-1,3-ditiano y proponer una aproximación sistemática al estudio del efecto anomérico y el análisis
conformacional en sulfuros orgánicos que contengan el grupo difenílfosfinoilo mediante el empleo de la teoría de átomos en moléculas.11
Para esto, se realizó la optimización geométrica a nivel B3LYP/6-31G(d,p) de los confórmeros relevantes del
1-difenilfosfinoil-3-tiometilpropano y sus correspondientes sulfóxido y sulfona, estudiando también la topología de su densidad electrónica en el marco
de la teoría de átomos en moléculas con la finalidad de caracterizar los enlaces presentes en dichas moléculas. Además de lo anterior, se emprendió el
cálculo y análisis de la energía, la población electrónica, el dipolo y la carga de todos los átomos de los confórmeros relevantes del
2-dimetilfosfinoil-1,3- ditiano y el dimetilfosfinoil(metansulfanil)-metano. Las contribuciones atómicas (W) a las propiedades moleculares se
calcularon mediante integración de la densidad de dicha propiedad en el volumen del contenedor atómico empleando la teoría de átomos en moléculas.
TEORÍA DE ÁTOMOS EN MOLÉCULAS
La teoría de átomos en moléculas (AIM por sus siglas en inglés), desarrollada por Richard F.W. Bader,12 proporciona una descripción rigurosa del enlace
y del átomo, basados por primera vez en la mecánica cuántica, por lo que es posible derivar conceptos útiles en química experimental tales como
aromaticidad, grupos funcionales, deslocalización, etc., a partir de la mecánica cuántica, mediante el estudio topológico12 de la densidad electrónica
r(r) del sistema en estudio.
Debido a que los núcleos tienen la propiedad fundamental de ser atractores en el campo vectorial del gradiente de la densidad electrónica ¾r( r), el espacio dominado por éstos está separado en una sección del espacio real que se denomina contenedor atómico. Estas regiones
dividen a la molécula en el concepto clásico de átomos. Un átomo está definido como la unión de un atractor y su contenedor atómico asociado. La
estructura de una molécula es definida sin ambigüedad determinando los puntos críticos a lo largo de la densidad electrónica, los cuales corresponden a
puntos donde el gradiente de la densidad electrónica es igual a cero (¾ r = 0).
¾r es una vector en la dirección de incremento de r(r). Una curvatura negativa en dichos puntos indica que la densidad es un máximo, y
si es positiva es un mínimo. !Asociadas a dicho PC existen una serie de trayectorias de ¾r"(r)# que inician en el infinito y terminan
en el PC; éstás determinan una superficie interatómica que separa a los contenedores de átomos vecinos.13
Si un par de núcleos comparten r(r) (enlazados en términos clásicos), existen además un par de trayectorias únicas que se inician en
dicho punto crítico y terminan en los átomos vecinos, a través de una trayectoria que es máxima en r(r), dicho punto crítico se denomina punto crítico
de enlace (PCE). Para una geometría en equilibrio existen una serie de líneas donde r(r) es máximo, que se denominan trayectorias de enlace, debido a
que si se trazan las uniones de los núcleos asociadas a dichas trayectorias de enlace (gráfica molecular) se reproducen los enlaces químicos que son
asignados bajo consideraciones clásicas. La densidad en el PCE es directamente proporcional con la fuerza del enlace. Un PCE es un punto de silla en la
densidad electrónica, ya que es un máximo en la dirección de la superficie que divide a los contenedores atómicos (Línea de superficie interatómica), y
un mínimo en la dirección de los núcleos. Las líneas que se originan y terminan en los núcleos se conocen como líneas de interacción atómica (LIA) o
trayectorias de enlace. Con lo anterior un enlace se define como: “Dos átomos están enlazados cuando comparten una SIA, exista un PCE y una LIA entre
ellos”.
INTEGRACIÓN ATÓMICA
Cada átomo en AIM está delimitado por una superficie que cumple la condición de cero flujo siguiente: ¾r"(r)# . n ( r )= 0 ² r S. Una propiedad atómica A (W) está definida como
una integral de volumen en la región del espacio dominada por el contenedor atómico de ese núcleo:
Á(Ù)
= Ú dôôa(r)
Ù
Donde a( r ) es la densidad de la propiedad a estudiar, como la densidad de carga r"(r)# y la densidad del momento dipolar rW¾r"(r)#. La población electrónica N(W) se relaciona con el momento monopolar de la siguiente
manera: N(W) = - Q00(W). El volumen atómico se define
como v(W) = Ú dt
W
y la energía atómica como
E
(W) = (1+ g )Ú K (r)dt
W
Donde K(r) es la densidad de la energía cinética y g es el virial < V > /< T > de la función de onda. El error de
integración L (W) mide la diferencia entre la densidad de la energía cinética K(r) y G( r); L(W) = K(W) - G(W). Para obtener valores confiables de integración L(W) tener un valor máximo
de 4x 10-4 ua.
OBJETIVOS Y METODOLOGÍA OBJETIVOS
El presente trabajo tiene el siguiente objetivo general:
El estudio de interacciones electrostáticas del tipo C-H --- O-P presentes en el 2- difenilfosfinoil-1,3-ditiano, sus análogos cíclicos y de cadena
abierta con la finalidad de explicar el origen de la preferencia conformacional de dichos sistemas. Para lo cual se establecieron los siguientes
objetivos particulares, de acuerdo a la metodología empleada:
Teoría
de
Funcionales
de
la densidad electrónica
-
Determinación de los parámetros geométricos y energía de los confórmeros relevantes del dimetilfosfinoil-3-tiometilpropano a nivel de teoría
Becke3LYP/6-31G(d,p).
-
Obtención de la función de onda de todos los confórmeros anteriores
Teoría
de
Átomos en Moléculas
· Realizar el estudio topológico de la densidad electrónica de los confórmeros relevantes del
dimetilfosfinoil-3-tiometilpropano,2-dimetilfosfinoil-1,3-ditiano y el dimetilfosfinoil(metansulfanil)-metano obteniendo las propiedades de todos los
puntos críticos de dichos sistemas.
Verificar si es posible establecer un modelo que explique la barrera rotacional del 2- dimetilfosfinoil-1,3-ditiano y el
dimetilfosfinoil(metansulfanil)-metano a partir de la densidad electrónica en el punto crítico de enlace, la energía E(W), la población electrónica
N(W), el dipolo y la carga Q(W) atómica.
MÉTODOS COMPUTACIONALES
Se realizó la optimización completa, el cálculo de frecuencias y el análisis termoquímico de todos los compuestos de interés a nivel B3LYP/6-31G(d,p)
en el programa14 Gaussian 94, Versión DEC-AXF-OSF/1-G94, de donde se obtuvo la función de onda que es requerida por el paquete AIMPAC15 para
obtener las propiedades atómicas integradas: Energía E(W), Población electrónica N(W), Carga Q(W) y Error de integración L(W), así como las propiedades
de los puntos críticos de la densidad electrónica: densidad (r), Laplaciano (¾2 r) y elipticidad (e). Los
puntos críticos se calcularon en el programa Extreme del paquete AIMPAC, las propiedades atómicas se obtuvieron con el programa PROAIM del mismo paquete. Los cálculos ab initio que aquí se presentan, fueron realizados en una computadora Digital Unix Versión
DEC-AXF-OSF perteneciente a la Escuela de Química de la Universidad La Salle, México. El análisis de la topología de la densidad electrónica se realizó
en tres computadoras SGI con sistema operativo Irix 6.5.13. también de la Universidad La Salle. Las moléculas se graficaron en los programas Molecule, Viewer Pro y Molden.
RESULTADOS Y DISCUSIÓN
ANÁLISIS CONFORMACIONAL DEL DIMETILFOSFINOIL(METANSULFANIL)-METANO
Se realizó la optimización geométrica y se obtuvo la función de onda de tres confórmeros gauche y tres confórmeros anti del
dimetilfosfinoil(metansulfanil)-metano (DMF-MSM Esquema 1) que resultan del giro del enlace C-P. Todas las estructuras son mínimos,
sin eigenvalores negativos ni frecuencias imaginarias. La geometría de estas moléculas ya ha sido reportada con anterioridad,11 de donde se establece
que no existe evidencia de interacciones estéreo
electrónicas del tipo nS Æ s*C-P. Se esperaría que 1“-g fuera un confórmero de baja energía si
tales interacciones existieran, por el contrario, 1-g donde el átomo de oxígeno apunta en dirección del grupo tiometilo y el enlace P-O es
antiperiplanar al enlace C-S es más estable por
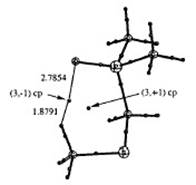
3.9 Kcal/mol. Como ya ha sido descrito,11 el confórmero 1-g presenta una interacción electrostática estabilizante entre el oxígeno y un hidrógeno a al
átomo de azufre (Figura 2), la
cual ha sido caracterizada por medio del estudio topológico de sus propiedades locales en la densidad electrónica, sin embargo, no ha sido reportado el
análisis de las propiedades integradas para tales confórmero.
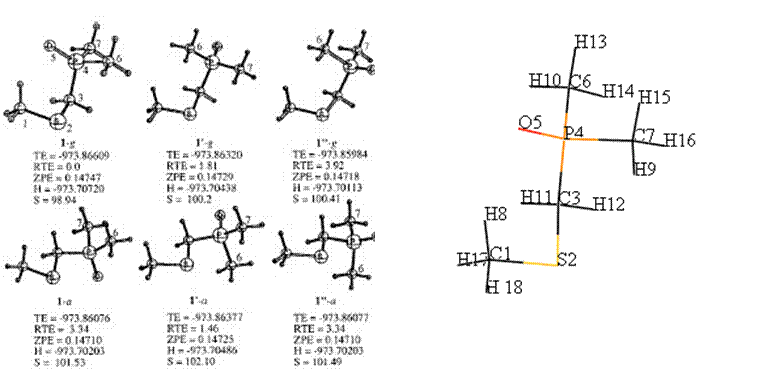 Esquema
1.
Energía total (TE, hartrees), Energía total relativa (TRE Kcal/mol), Energía del punto Cero (ZPE, hartrees), Entalpía (H, hartrees) y Entropía (S eu)
del DMF-MSM.
Esquema
1.
Energía total (TE, hartrees), Energía total relativa (TRE Kcal/mol), Energía del punto Cero (ZPE, hartrees), Entalpía (H, hartrees) y Entropía (S eu)
del DMF-MSM.
NUMERACIÓN ASIGNADA
Dicho estudio llevaría a un mejor entendimiento del efecto anomérico, ya que la energía de los átomos depende, además de su conectividad de su
conformación, sin embargo, sólo existen algunos estudios reportados en la literatura que establecen cuál es la evolución de la energía de los átomos de
una molécula cuando existen cambios conformacionales.16 En este trabajo se analiza la contribución de la energía y población atómicas a la molecular
cuando existen cambios
conformacionales en derivados del 2-difenilfosfinoil-1,3-ditiano. En la Tabla 1 se presenta la evolución de la energía atómica en los confórmeros del
esquema 1 con la numeración que será utilizada para designar a cada uno de sus átomos.
Figura
2.
Puntos críticos relevantes en 1-g
Un parámetro que ha demostrado ser importante para determinar la confiabilidad de la integración atómica es la diferencia entre la sumatoria de las
energías atómicas SW E(W) y la energía molecular calculada por el método de campo auto consistente (SCF, por sus siglas en inglés) al nivel de teoría
en el cual se obtuvo la función de onda,17 dicha diferencia es causada por el error de integración numérica y debe ser menor de 1. 0 Kcal. mol-1. En la
tabla 1 se presentan los resultados de E(SCF) - SW E(W) siendo el confórmero con menor diferencia el 1-g con 0.00349 Hartress.
La mayor diferencia se presenta en el confórmero 1’-g. Dado que los datos obtenidos son confiables, se determinó la energía del grupo dimetilfosfinoilo
y la cadena derivada del dimetilsulfuro, con la finalidad de encontrar cuál es la evolución energética de cada segmento debido a la rotación interna.
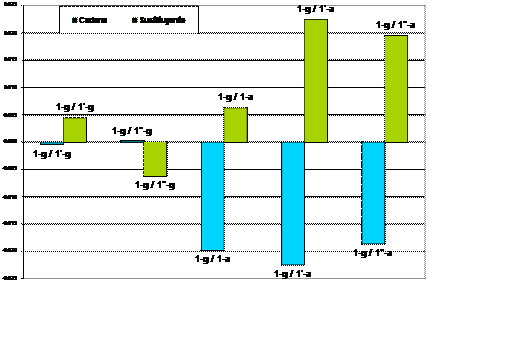
En la Gráfica 1 se presenta la diferencia de la energía del grupo y de la cadena, tomando como referencia al confórmero 1-g. Por ejemplo, la energía
del sustituyente
en 1-g es de -495.91475 Hartrees y en 1’-g de -495.92467Hartrees, si realizamos la diferencia, resulta que el sustituyente es –7.42 Kcal mol-1 más
estable en 1’-g, por lo que la barra en la gráfica adopta valores negativos.18
Tabla
1
. Evolución de la Energía atómica ( E (W), Hartrees) de todos los confórmeros del
DMF-MSM
a nivel B3LYP/6-31G(d,p)
|
Atomo
|
E(
W
)
1-g
|
E(
W
)
1 '-g
|
E(
W
)
1''-g
|
E
(
W
)
1
-
a
|
E
(
W
)
1
'-a
|
E
(
W
)
1
''-a
|
|
C 1
|
-37.83301
|
-37.82272
|
-37.82352
|
-37.82261
|
-37.82134
|
-37.82254
|
|
S 2
|
-398.91483
|
-398.91406
|
-398.90080
|
-398.90207
|
-398.92188
|
-398.90208
|
|
C 3
|
-38.13135
|
-38.12895
|
-38.12686
|
-38.12958
|
-38.13093
|
-38.12969
|
|
P 4
|
-340.35355
|
-340.35427
|
-340.35204
|
-340.34047
|
-340.33815
|
-340.34080
|
|
O 5
|
-75.61269
|
-75.60763
|
-75.61317
|
-75.61394
|
-75.60940
|
-75.61390
|
|
C 6
|
-38.13008
|
-38.14058
|
-38.13810
|
-38.13911
|
-38.14214
|
-38.13199
|
|
C 7
|
-38.14170
|
-38.13759
|
-38.12880
|
-38.13205
|
-38.14223
|
-38.13902
|
|
H 8
|
-0.59746
|
-0.61400
|
-0.62631
|
-0.62439
|
-0.62096
|
-0.62241
|
|
H 9
|
-0.60406
|
-0.62066
|
-0.61196
|
-0.61146
|
-0.61088
|
-0.60520
|
|
H 10
|
-0.61183
|
-0.61184
|
-0.60738
|
-0.60521
|
-0.61087
|
-0.61143
|
|
H 11
|
-0.61465
|
-0.61003
|
-0.61967
|
-0.62327
|
-0.61438
|
-0.61674
|
|
H 12
|
-0.61706
|
-0.61025
|
-0.60966
|
-0.61673
|
-0.61442
|
-0.62325
|
|
H 13
|
-0.61044
|
-0.61253
|
-0.61079
|
-0.61167
|
-0.61208
|
-0.61050
|
|
H 14
|
-0.61942
|
-0.61219
|
-0.62183
|
-0.62020
|
-0.61303
|
-0.62000
|
|
H 15
|
-0.61136
|
-0.61133
|
-0.61064
|
-0.61050
|
-0.61206
|
-0.61161
|
|
H 16
|
-0.61961
|
-0.61604
|
-0.62094
|
-0.62003
|
-0.61307
|
-0.62020
|
|
H 17
|
-0.62460
|
-0.61452
|
-0.62339
|
-0.62241
|
-0.62097
|
-0.62439
|
|
H 18
|
-0.61839
|
-0.62499
|
-0.61441
|
-0.61508
|
-0.61465
|
-0.61508
|
|
Total
|
-973.86610
|
-973.86419
|
-973.86027
|
-973.86077
|
-973.86344
|
-973.86081
|
|
Mol
|
-973.86609
|
-973.86320
|
-973.85984
|
-973.86076
|
-973.86377
|
-973.86077
|
|
Delta
|
0.00349
|
0.61962
|
0.27019
|
0.00508
|
-0.20533
|
0.03012
|
|
Cadena
|
-477.95135
|
-477.93952
|
-477.94462
|
-477.95614
|
-477.95952
|
-477.95617
|
|
Sustituyente
|
-495.91475
|
-495.92467
|
-495.91565
|
-495.90463
|
-495.90392
|
-495.90465
|
|
H8-C1-S-C3-P-O
|
-891.44289
|
-891.44163
|
-891.44270
|
-891.43307
|
-891.44265
|
-891.43141
|
Como puede concluirse de la tabla 1, la conformación observada es un balance entre la estabilización del sustituyente (POMe2) y la cadena, siendo más
estable el primero en la conformación gauche y la cadena en la conformación anti. Los confórmeros 1’-g y 1’-a presentan un arreglo donde el enlace P-O
es antiperiplanar al enlace S2-C3, donde posiblemente existe una interacción estereoelectrónica del tipo nO s*C-P la cual estabiliza a dichos
confórmeros, sin embargo, el puente de hidrógeno antes mencionado parece ser aún más importante, ya que el
mínimo global observado es 1-g, lo cual coincide con los datos experimentales reportados.10
La evolución energética de los núcleos de DMF-MSM al pasar del confórmero 1-g al 1’-a muestra que la mayor estabilidad del
sustituyente en 1-g está regida por el átomo de fósforo, y que la mayor estabilidad de la cadena en 1’-a es gobernada por el H8 el cual participa en
una interacción electrostática con el grupo dimetilfosfinoilo (figura 1). Contrario a lo esperado, el átomo de oxígeno presenta una variación en
energía mucho menor a la de los núcleos antes mencionados cuando una interacción nO s*C-P lo estabiliza. La transferencia de carga de un núcleo a
otro puede observarse al calcular la población electrónica. En la tabla 2 se presenta la integración atómica de la población electrónica para los
confórmeros de DMF-MSM, al igual que en E (W) la diferencia entre la sumatoria de la propiedad atómica y la global esperada es un
parámetro de confiabilidad del cálculo, como puede verse de dicha tabla, los valores obtenidos son confiables de acuerdo con lo reportado en la
literatura.19
|
1-g
/ 1'-g
|
Cadena
Sustituyente
|
1-g
/ 1'-a
|
|
|
|
|
|
|
|
|
|
|
1-g
/ 1''-g
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1-g
/ 1''-g
|
|
|
1-g
/ 1-a
|
1-g
/ 1''-a
|
|
1-g
/ 1'-a
|
|
1-g
/ 1'-g
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
0.015
Gráfica1.
Comparación de la estabilización de la cadena y el sustituyente tomando como referencia el 1-g
0.010
1-g
/ 1-a
1-g
/ 1''-a
0.005
0.000
-0.005
-0.010
-0.015
Gráfica
3.
Comparación de la población electrónica de la cadena y el sustituyente tomando como referencia el 1-g
El átomo de fósforo presenta una mayor población electrónica en los confórmeros gauche y H8 presenta una población muy baja en 1-g con respecto a los
demás confórmeros, lo cual es debido a la participación en un puente de hidrógeno.
Se calculó también N (W) para el grupo dimetilfosfinoilo y la cadena, en la gráfica 3 se presenta la evolución de dicha propiedad con el cambio
conformacional. La población electrónica sigue la misma tendencia que la energía cuando se compara el confórmero 1-g con los confórmeros anti, siendo
la mas alta con 1’-a. Tal comportamiento es debido a una interacción nO s*C-P favorecida por la disposición antiperiplanar entre los enlaces P-O y
S2-C3. en 1’-a.
Tabla
2
. Evolución de la Población electrónica ( N (W), electrones) a nivel B3LYP/6-31G(d,p) de todos los confórmeros del DMF-MSM
|
Atomo
|
N
(
W
)
1
-
g
|
N
(
W
)
1
'
-
g
|
N
(
W
)
1
''
-
g
|
N
(
W
)
1
-
a
|
N
(
W
)
1
'-a
|
N
(
W
)
1
''-a
|
|
C 1
|
6.02827
|
6.01879
|
6.01992
|
6.02109
|
6.01659
|
6.01988
|
|
S 2
|
16.00459
|
16.00416
|
15.96762
|
15.96506
|
16.01221
|
15.96496
|
|
C 3
|
6.64051
|
6.63324
|
6.63430
|
6.63269
|
6.63256
|
6.63281
|
|
P 4
|
11.87825
|
11.87695
|
11.88704
|
11.86979
|
11.86414
|
11.86850
|
|
O 5
|
9.48150
|
9.47426
|
9.47503
|
9.47262
|
9.47464
|
9.47260
|
|
C 6
|
6.58809
|
6.60132
|
6.59541
|
6.61071
|
6.60225
|
6.58774
|
|
C 7
|
6.60281
|
6.59301
|
6.58538
|
6.58766
|
6.60134
|
6.59878
|
|
H 8
|
0.92493
|
0.98673
|
0.99888
|
0.99888
|
0.98976
|
0.99329
|
|
H 9
|
0.96320
|
0.98896
|
0.98246
|
0.98089
|
0.97955
|
0.96526
|
|
H 10
|
0.98187
|
0.98231
|
0.96927
|
0.96523
|
0.97955
|
0.98085
|
|
H 11
|
0.97536
|
0.96471
|
0.98739
|
0.99401
|
0.97256
|
0.97743
|
|
H 12
|
0.98142
|
0.96214
|
0.95996
|
0.97740
|
0.97265
|
0.99398
|
|
H 13
|
0.97921
|
0.98398
|
0.97941
|
0.98165
|
0.98282
|
0.97914
|
|
H 14
|
1.00113
|
0.98322
|
1.00699
|
1.00264
|
0.98458
|
1.00223
|
|
H 15
|
0.98117
|
0.98069
|
0.97941
|
0.97912
|
0.98274
|
0.98155
|
|
H 16
|
1.00163
|
0.98971
|
1.00479
|
1.00224
|
0.98476
|
1.00271
|
|
H 17
|
0.99917
|
0.97789
|
0.99599
|
0.99327
|
0.98980
|
0.99889
|
|
H 18
|
0.98792
|
0.99492
|
0.97780
|
0.97963
|
0.97859
|
0.97963
|
|
Total
|
74.00103
|
73.99697
|
74.00703
|
74.01457
|
74.00109
|
74.00021
|
|
Mol
|
74.00000
|
74.00000
|
74.00000
|
74
|
74.00000
|
74
|
|
Delta
|
-0.00103
|
0.00303
|
-0.00703
|
-0.01457
|
-0.00109
|
-0.00021
|
|
Cadena
|
33.54217
|
33.54257
|
33.54186
|
33.56202
|
33.56472
|
33.56087
|
|
Sustituyente
|
40.45886
|
40.45440
|
40.46517
|
40.45255
|
40.43637
|
40.43935
|
|
S-C-P-O
|
50.95804
|
50.99413
|
50.98278
|
50.96011
|
50.98990
|
50.95203
|
En el caso de los derivados del 1,3-ditiano, todos los isómeros mantienen un arreglo de silla.
En el esquema 2 se muestra que el confórmero 2-ax es 2.25 kcal mol-1 más estable que 2-eq que es el confórmero observado experimentalmente, el giro del
enlace C-P al pasar de 1-ax a 1’-
ax desestabiliza el sistema por 6.37 kcal mol-1, si n
s*C-P
fuera una interacción estéreo
electrónica dominante la diferencia debería ser mucho menor, debido únicamente a la repulsión estérica de los metilos del grupo dimetilfosfinoilo con
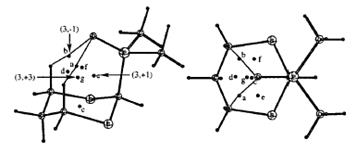
los hidrógenos 4-6 syn-diaxiales del anillo, por lo que debe existir otro factor dominante en 1-ax, tal factor es el puente de hidrógeno que provoca
que el sistema adopte una configuración similar a la del adamantano (Figura 3), el cual
ya fue caracterizado topológicamente11 mediante la Teoría de átomo en moléculas.
Figura
3.
Puntos críticos relevantes en 1-ax asociados con interacciones débiles en vistas diferentes
Esquema
2.
Energía total (TE, hartrees), Energía total relativa (TRE Kcal/mol), energía del puntoCero (ZPE, hartrees), Entalpía (H, hartrees) y Entropía (S eu)
del dimetilfosfinoil-1,3-ditiano
Se determinaron las propiedades atómicas de todos los átomos del 2-dimetilfosfinoil-1,3- ditiano, las cuales se presentan en la tabla 3, el error de
integración es suficientemente pequeño por lo que los valores obtenidos son confiables. Se evaluó la energía del anillo 1,3-ditiano y del sustituyente
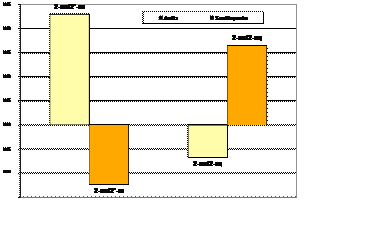
dimetilfosfinoilo en los confórmeros 1-ax, 1’-ax y 1-eq por ser estos los confórmeros observados experimentalmente. En la Gráfica 3 se muestra que el
anillo se ve estabilizado al
pasar del confórmero 2-ax al 2-eq aproximadamente 5 kcal mol-1, sin embargo, este mismo se desestabiliza en el confórmero 2’-ax por casi 15 kcal mol-1,
el sustituyente por el contrario, muestra una gran estabilización al pasar del confórmero 2-ax al 2’-ax de 8 kcal mol -1, este
mismo se ve desestabilizado por casi 10 kcal mol-1 en el confórmero 1-eq con respecto a 1-ax. La evolución energética del anillo y del sustituyente
está regida por los núcleos que se desestabilicen más al pasar de una conformación a otra. En la tabla 4 se muestra que los hidrógenos 11 y 12 se ven
estabilizados en los confórmeros 2-eq y 2’-ax, el comportamiento de los átomos de azufre y del carbono anomérico es contrario. En contra de lo esperado
el átomo de oxígeno no tiene una participación relevante en la evolución energética del sustituyente, al igual que los carbonos 9 y 10, los cuales solo
varían levemente su energía al pasar de 2-ax a 2’- ax, esos mismo núcleos prácticamente no varían su energía al pasar de 2-ax a 1-eq. Como puede
observarse la evolución energética del 2-dimetilfosfinoil-1,3-ditiano a favor del confórmero 1-ax está controlada por los átomos de azufre, el carbono
C2 y él átomo de fósforo.
Tabla
4.
Átomos que rigen la evolución energética (Kcal /mol) en los derivados del 1,3-ditiano tomando como referencia a 1-ax
|
Atomo
|
2-ax/2'-ax
|
2-ax/2-eq
|
|
S 1
|
15.47501
|
5.24974
|
|
C 2
|
11.51116
|
3.02566
|
|
S 3
|
3.57053
|
4.14909
|
|
P 7
|
-5.02259
|
10.39156
|
|
O 8
|
1.07022
|
0.14790
|
|
H 11
|
-14.50626
|
-13.05950
|
|
H 12
|
-15.07031
|
-13.04756
|
Tabla
3
. Evolución de la Energía atómica ( E (W), Hartees) de los confórmeros 1-ax, 1’-ax y 1-eq del 2-dimetilfosfinoil-1,3-ditiano a nivel
B3LYP/6-31G(d,p)
|
Atomo
|
E
(
W
)
2
-ax
|
E
(
W
)
2
'-ax
|
E
(
W
)
2-eq
|
|
S 1
|
-398.84920
|
-398.82454
|
-398.84083
|
|
C 2
|
-38.10896
|
-38.09062
|
-38.10414
|
|
S 3
|
-398.84925
|
-398.84356
|
-398.84264
|
|
C 4
|
-37.83899
|
-37.83441
|
-37.83068
|
|
C 5
|
-37.80909
|
-37.81341
|
-37.81126
|
|
C 6
|
-37.83891
|
-37.83522
|
-37.83097
|
|
P 7
|
-340.29306
|
-340.30106
|
-340.27650
|
|
O 8
|
-75.59799
|
-75.59629
|
-75.59776
|
|
C 9
|
-38.13006
|
-38.13172
|
-38.12973
|
|
C 10
|
-38.13039
|
-38.12603
|
-38.12983
|
|
H 11
|
-0.61132
|
-0.63444
|
-0.63213
|
|
H 12
|
-0.61130
|
-0.63531
|
-0.63209
|
|
H 13
|
-0.62795
|
-0.62405
|
-0.62463
|
|
H 14
|
-0.62796
|
-0.62445
|
-0.62459
|
|
H 15
|
-0.63580
|
-0.63619
|
-0.63662
|
|
H 16
|
-0.63338
|
-0.62917
|
-0.62844
|
|
H 17
|
-0.61425
|
-0.60804
|
-0.62406
|
|
H 18
|
-0.61145
|
-0.61005
|
-0.61866
|
|
H 19
|
-0.61896
|
-0.62046
|
-0.61138
|
|
H 20
|
-0.60433
|
-0.61344
|
-0.60524
|
|
H 21
|
-0.61148
|
-0.61191
|
-0.61927
|
|
H 22
|
-0.61896
|
-0.61169
|
-0.61113
|
|
H 23
|
-0.60429
|
-0.61182
|
-0.60517
|
|
Total
|
-1449.47730
|
-1449.46787
|
-1449.46774
|
|
EnergíaSCF
|
-1449.47687
|
-1449.46671
|
-1449.46758
|
|
Diferencia
|
0.27404
|
0.72545
|
0.10187
|
|
Anillo
|
-953.65635
|
-953.63340
|
-953.66308
|
|
Sustituyente
|
-495.82096
|
-495.83447
|
-495.80466
|
|
H11-C6-S1-C-P-O
|
-891.29944
|
-891.28216
|
-891.28233
|
|
H8-C6-S3-C-P-O
|
-891.29955
|
-891.30125
|
-891.28380
|
De acuerdo con la simetría de 1-ax se esperaría que la variación en energía de sus núcleos tuvieran un comportamiento también simétrico, sin embargo,
los átomos de azufre se desestabilizan en diferente proporción al pasar de 1-ax a 1’-ax, siendo S3 el menos desestabilizado, lo que es congruente con
una interacción ns s*C-P, la cual parece no ser tan importante como el puente de hidrógeno antes descrito, la integración de la población electrónica
da evidencia de tales interacciones. La gráfica 3 muestra que al igual que en el DMF-MSM la población electrónica en los confórmeros
2-ax, 2’-ax y 2-eq muestra la misma tendencia que la energía si se toma como referencia a 1-ax, de donde se puede concluir que el aumento de la
población electrónica estabiliza al anillo o al sustituyente si éstos son capaces de deslocalizarla. Del mismo modo que para la energía, la evolución
de la población electrónica está regida por los hidrógenos 11 y 12 a favor de 2-eq, mientras que la tendencia a favor de 2-ax sobre 2-eq es dada por
todos los núcleos pesados excepto el C5 que prácticamente no varía su N(W) (Tabla 5). En el caso de 1’-ax la situación es más errática debido a la
existencia interacciones electrostáticas y estéreo electrónicas competitivas.
En el apéndice 1 de este trabajo se presentan las propiedades integradas Q(W) y L(W) de todos los núcleos de los confórmeros que han sido reportados.
Gráfica 3.
Comparación de la población electrónica del anillo y el sustituyente tomando como referencia el 2-ax
 -
-
-
Tabla
5
. Evolución de la población electrónica (N (W), electrones) a nivel B3LYP/6-31G(d,p) de todos los confórmeros 1-ax, 1’-ax y 1-eq del 2-dimetilfosfinoil-1,3-ditiano
|
Atomo
|
N
(
W
)
2
-ax
|
N
(
W
)
2
'-ax
|
N
(
W
)
2-eq
|
|
S 1
|
16.00760
|
15.95391
|
15.97523
|
|
C 2
|
6.69443
|
6.68019
|
6.68439
|
|
S 3
|
16.00756
|
16.00391
|
15.97986
|
|
C 4
|
6.01699
|
6.01044
|
6.01306
|
|
C 5
|
5.89288
|
5.89233
|
5.89511
|
|
C 6
|
6.01699
|
6.01124
|
6.01383
|
|
P 7
|
11.91498
|
11.94038
|
11.89710
|
|
O 8
|
9.48344
|
9.46965
|
9.46602
|
|
C 9
|
6.61220
|
6.64768
|
6.59542
|
|
C 10
|
6.59830
|
6.59027
|
6.59609
|
|
H 11
|
0.94766
|
1.00894
|
1.00944
|
|
H 12
|
0.94751
|
1.00985
|
1.00920
|
|
H 13
|
0.99602
|
0.98548
|
0.98786
|
|
H 14
|
0.99605
|
0.98627
|
0.98776
|
|
H 15
|
1.01868
|
1.01940
|
1.02107
|
|
H 16
|
1.01195
|
1.00176
|
0.99898
|
|
H 17
|
0.96100
|
0.94068
|
0.98163
|
|
H 18
|
0.98173
|
0.97731
|
0.99889
|
|
H 19
|
1.00044
|
0.99901
|
0.98118
|
|
H 20
|
0.96262
|
0.98205
|
0.96395
|
|
H 21
|
0.98178
|
0.98263
|
1.00043
|
|
H 22
|
1.00042
|
0.98443
|
0.98061
|
|
H 23
|
0.96266
|
0.98258
|
0.96409
|
|
Total
|
104.01390
|
104.06038
|
104.00121
|
|
Molecular
|
104
|
104
|
104
|
|
Diferencia
|
-0.01390
|
-0.06038
|
-0.00121
|
|
Anillo
|
63.51532
|
63.50439
|
63.55743
|
|
Sustituyente
|
40.49858
|
40.55600
|
40.44378
|
|
H11-C6-S1-C-P-O
|
51.06510
|
51.06431
|
51.04601
|
|
H8-C6-S3-C-P-O
|
51.06492
|
51.11440
|
51.04963
|
CONCLUSIONES
Se logró realizar la integración numérica de las propiedades atómicas de los confórmeros relevantes del dimetilfosfinoil-metansulfanil-metano y del
2-dimetilfosfinoil-1,3-ditiano, obteniendo resultados confiables de acuerdo con lo reportado en la literatura.
Los resultados aquí presentados permiten concluir que la conformación observada experimentalmente es resultado de una competencia entre la
estabilización del sustituyente y de la cadena / anillo. En el caso del dimetilfosfinoil-metansulfanil-metano y del 2-dimetilfosfinoil-1,3- ditiano la
transferencia de densidad electrónica de la cadena hacia el átomo de fósforo mediante
una interacción electrostática del tipo C-H --- O-P y la capacidad del sustituyente para estabilizarla a costa de la cadena / anillo es el origen de la
preferencia conformacional del sistema estudiado, lo que explica el porqué el modelo hiperconjugativo nS Æ s*C-P no es capaz de predecir las geometrías
y energías obtenidas experimentalmente, si acaso existiese alguna interacción estéreo electrónica sería sólo del tipo nO Æ s*C-P la cual es consistente
con los resultados aquí presentados y otros reportados en la literatura. Como perspectiva a futuro, queda
todavía realizar el cálculo de otros sulfuros orgánicos que contengan el grupo dimetilfosfinoil, además de la comprobación experimental del modelo
propuesto en un derivado no cíclico donde no existe el arreglo S-C-P-O y que además sea capaz de formar un puente de hidrógeno, ambas líneas están
siendo exploradas por los autores.
APÉNDICE 1.
CARGA Q(W) Y ERROR DE INTEGRACIÓN L(W) CALCULADOS PARA TODOS LOS NÚCLEOS DE TODOS LOS CONFÓRMEROS DEL DMF-MSM
|
Atomo
|
q
(
W
)
1
-
g
|
L
(
W
)
1-g
|
q
(
W
)
1
'
-
g
|
L
(
W
)
1'-g
|
q
(
W
)
1
''
-
g
|
L
(
W
)
1''-g
|
q
(
W
)
1
-
a
L
(
W
)
1
-
a
q
(
W
)
1
'-a L
(
W
)
1
'-a
q
(
W) 1
''-a
|
L
(
W
)
1
''-a
|
|
C 1
|
-0.02827
|
0.00020
|
-0.01879
|
-0.00001
|
-0.01992
|
0.00187
|
-0.02109
|
0.00004
|
-0.0166
|
0.00001
|
-0.01988
|
0.00087
|
|
S 2
|
-0.00459
|
0.00019
|
-0.00416
|
-0.00017
|
0.03238
|
-0.00018
|
0.03494
|
-0.00044
|
-0.0122
|
-0.000055
|
0.03504
|
-0.00048
|
|
C 3
|
-0.64051
|
-0.00013
|
-0.63324
|
-0.00013
|
-0.63430
|
-0.00047
|
-0.63269
|
0.00023
|
-0.6326
|
-0.00096
|
-0.63281
|
0.00023
|
|
P 4
|
3.12176
|
-0.00136
|
3.12305
|
0.00130
|
3.11296
|
-0.00784
|
3.13021
|
-0.00329
|
3.1359
|
-0.000631
|
3.13150
|
-0.00207
|
|
O 5
|
-1.48150
|
0.00007
|
-1.47426
|
0.00008
|
-1.47503
|
0.00009
|
-1.47262
|
0.00007
|
-1.4746
|
0.00007
|
-1.47260
|
0.00006
|
|
C 6
|
-0.58809
|
-0.00014
|
-0.60132
|
-0.00021
|
-0.59541
|
0.00066
|
-0.61071
|
-0.00901
|
-0.6023
|
-0.000476
|
-0.58774
|
0.00072
|
|
C 7
|
-0.60281
|
-0.00018
|
-0.59301
|
0.00164
|
-0.58538
|
0.00022
|
-0.58766
|
0.00079
|
-0.6013
|
0.00034
|
-0.59878
|
-0.00050
|
|
H 8
|
0.07507
|
0.00003
|
0.01327
|
0.00008
|
0.00112
|
0.00008
|
0.00112
|
0.00008
|
0.0102
|
0.00006
|
0.00671
|
0.00008
|
|
H 9
|
0.03680
|
0.00014
|
0.01104
|
0.00006
|
0.01754
|
0.00006
|
0.01911
|
0.00005
|
0.0205
|
0.00007
|
0.03474
|
0.00006
|
|
H 10
|
0.01813
|
0.00007
|
0.01769
|
0.00006
|
0.03073
|
0.00015
|
0.03477
|
0.00007
|
0.0205
|
0.00006
|
0.01915
|
0.00006
|
|
H 11
|
0.02464
|
0.00010
|
0.03529
|
0.00011
|
0.01261
|
0.00012
|
0.00599
|
0.00013
|
0.0274
|
0.00010
|
0.02257
|
0.00010
|
|
H 12
|
0.01858
|
0.00010
|
0.03786
|
0.00009
|
0.04004
|
0.00009
|
0.02260
|
0.00009
|
0.0274
|
0.00011
|
0.00602
|
0.00013
|
|
H 13
|
0.02079
|
0.00004
|
0.01602
|
0.00006
|
0.02059
|
0.00006
|
0.01835
|
0.00006
|
0.0172
|
0.00006
|
0.02086
|
0.00007
|
|
H 14
|
-0.00113
|
0.00007
|
0.01678
|
0.00003
|
-0.00699
|
0.00009
|
-0.00264
|
0.00007
|
0.0154
|
0.000018
|
-0.00223
|
0.00006
|
|
H 15
|
0.01883
|
0.00005
|
0.01931
|
0.00006
|
0.02059
|
0.00006
|
0.02088
|
0.00006
|
0.0173
|
0.00007
|
0.01845
|
0.00006
|
|
H 16
|
-0.00163
|
0.00003
|
0.01029
|
0.00001
|
-0.00479
|
0.00007
|
-0.00224
|
0.00007
|
0.0152
|
0.00000
|
-0.00271
|
0.00005
|
|
H 17
|
0.00083
|
0.00006
|
0.02211
|
0.00005
|
0.00401
|
0.00007
|
0.00673
|
0.00008
|
0.0102
|
0.00007
|
0.00111
|
0.00008
|
|
H 18
|
0.01208
|
0.00005
|
0.00508
|
0.00008
|
0.02220
|
0.00005
|
0.02037
|
0.00005
|
0.0214
|
0.00005
|
0.02037
|
0.00005
|
|
Total
|
-0.00103
|
|
0.00303
|
|
-0.00703
|
-0.01457
|
|
-0.0011
|
|
-0.00021
|
|
Cadena
|
-0.54217
|
0.00060
|
-0.54257
|
0.00010
|
-0.54186
|
0.00164
|
-0.56202
|
0.00025
|
-0.56472
|
-0.00060
|
-0.56087
|
0.00104
|
|
Sust
|
0.54114
|
-0.00121
|
0.54560
|
0.00308
|
0.53483
|
-0.00638
|
0.54745
|
-0.01104
|
0.56363
|
-0.00042
|
0.56065
|
-0.00142
|
APÉNDICE 2.
CARGA Q(W) Y ERROR DE INTEGRACIÓN L(W) CALCULADOS PARA TODOS LOS NÚCLEOS LOS ISÓMEROS 2-ax, 2’-ax y 2-eq DEL 2-DIMETILFOSFINOIL-1,3-DITIANO
|
Atomo
|
q
(
W
)
2
-ax
|
L
(
W
)
2
-ax
|
q
(
W
)
2
'-ax
|
L
(
W
)
2
'-ax
|
q
(
W
)
2-eq
|
L
(
W
)
2-eq
|
|
S 1
|
-0.008
|
0.00014
|
0.046
|
0.00010
|
0.02478
|
0.00040
|
|
C 2
|
-0.694
|
0.00258
|
-0.680
|
0.00061
|
-0.68439
|
0.00067
|
|
S 3
|
-0.008
|
0.00012
|
-0.004
|
-0.00004
|
0.02014
|
0.00036
|
|
C 4
|
-0.017
|
-0.00008
|
-0.010
|
0.00227
|
-0.01306
|
-0.00033
|
|
C 5
|
0.107
|
0.00069
|
0.108
|
0.00097
|
0.10489
|
0.00059
|
|
C 6
|
-0.017
|
-0.00013
|
-0.011
|
0.00355
|
-0.01383
|
-0.00061
|
|
P 7
|
3.085
|
-0.00179
|
3.060
|
-0.01072
|
3.10290
|
-0.00242
|
|
O 8
|
-1.483
|
0.00004
|
-1.470
|
0.00008
|
-1.46602
|
0.00009
|
|
C 9
|
-0.612
|
-0.01085
|
-0.648
|
-0.03015
|
-0.59542
|
0.00003
|
|
C 10
|
-0.598
|
-0.00047
|
-0.590
|
-0.00101
|
-0.59609
|
-0.00034
|
|
H 11
|
0.052
|
0.00002
|
-0.009
|
-0.00005
|
-0.00944
|
0.00010
|
|
H 12
|
0.052
|
0.00009
|
-0.010
|
-0.00002
|
-0.00920
|
0.00010
|
|
H 13
|
0.004
|
0.00006
|
0.015
|
0.00007
|
0.01214
|
0.00006
|
|
H 14
|
0.004
|
0.00006
|
0.014
|
0.00007
|
0.01224
|
0.00007
|
|
H 15
|
-0.019
|
0.00005
|
-0.019
|
0.00007
|
-0.02107
|
0.00007
|
|
H 16
|
-0.012
|
0.00009
|
-0.002
|
0.00008
|
0.00102
|
0.00010
|
|
H 17
|
0.039
|
0.00012
|
0.059
|
0.00005
|
0.01837
|
0.00017
|
|
H 18
|
0.018
|
0.00006
|
0.023
|
0.00006
|
0.00111
|
0.00008
|
|
H 19
|
0.000
|
0.00007
|
0.001
|
0.00008
|
0.01882
|
0.00006
|
|
H 20
|
0.037
|
-0.00002
|
0.018
|
0.00002
|
0.03605
|
0.00006
|
|
H 21
|
0.018
|
0.00006
|
0.017
|
0.00006
|
-0.00043
|
0.00008
|
|
H 22
|
0.000
|
0.00007
|
0.016
|
-0.00286
|
0.01939
|
0.00006
|
|
H 23
|
0.037
|
-0.00012
|
0.017
|
0.00008
|
0.03591
|
0.00007
|
|
Total
|
-0.01390
|
-0.00911
|
-0.06038
|
-0.03664
|
-0.00121
|
-0.00048
|
|
Anillo
|
-0.51532
|
|
-0.50439
|
|
-0.55743
|
|
|
Sustituyente
|
0.50142
|
0.44400
|
|
0.55622
|
|
H11-C6-S1-C-P-O
|
0.93490
|
0.93569
|
|
0.95399
|
|
H8-C6-S3-C-P-O
|
0.93508
|
0.88560
|
|
0.95037
|
REFERENCIAS

1. Juaristi, E.; Valle, L.; Mora-Uzeta, C.; Valenzuela, B. A.; Joseph-Nathan, P.; Fredrich, M.F.J.Org. Chem. 47, 5038, 1982.
2. Scheleyer, P. V. R.; Jemmis, E. D.; Spitznagek, G.W. J. Am. Chem. Soc.,107, 6369, 1985.
3. Juaristi, E.; Valle, L.; Valenzuela, B. A.; Aguilar, M. A. J. Am. Chem. Soc., 108, 2000, 1986.
4. Juaristi, E.; Cuevas, G. The Anomeric Effect; CRC Press: Boca Raton, FL, Section 7.3. 1995.
5. Juaristi, E.; Valenzuela, B. A.; Valle, L.; McPhail, A. T. J. Org. Chem. 1984.
6. Juaristi, E.; Valle, L.; Valenzuela, B.A.; Aguilar, M.A.. J. Am. Chem. Soc., 108, 2000, 1986.
7. (a) Juaristi, E.; Tapia, J.; Méndez, R. Tetrahedron 42, 5038, 1986, (b) Arai, K.; Iwamura, H., Oki, M. Bull Chem Soc. Japan, 48,
3319, 1975.
8. Madrid, G.; Rochín. A; Juaristi, E.; Cuevas, G. J. Org. Chem., 66, 2925-2931, 2001.
9. Madrid, G.
Estudio experimental y computacional de los efectos estéricos, electrónicos y estéreo electrónicos que participan en la estabilidad conformacional
y en la reactividad de compuestos orgánicos
. Tesis Doctoral UNAM, 2001.
10. Cuevas, G. J. Am. Chem. Soc., 122, 692, 2000.
11. (a) Bader, R. F. W. Atoms in Molecules-a quantum theory; Clerendon Press: Oxford, 1990.
(b) R.F.W. Bader, Chem. Rev. 91, 893, 1991.
12. (a) Bader, R. F. W.; Carroll, M. T.; Cheeseman, J. R.; Chang, C. J. Am. Chem. Soc., 109, 7968, 1987, (b) Bader, R. F. W.; Essen,
H. J. Chem. Phys., 80, 1943, 1984, (c) Bader, R. F. W. J Phys. Chem. A, 102, 7314, 1998.
13. Popelier, Paul. Atoms in Molecules an introduction; Prentice Hall, Londres, 2000.
14. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Gill, P. M. W.; Johnson, B. G.; Robb, M. A.; Cheeseman, J. R.; Keith, T.; Petersson, G. A.;
Montgomery, J. A.; Raghavachari, K.; Al- Laham, M. A.; Zakrzewski, V. G.; Ortiz, J. V.; Foresman, J. B.; Cioslowski, J.; Stefanov, B. B.; Nanayakkara,
A.; Challacombe, M.; Peng, C. Y.; Ayala, P. Y.; Chen, W.; Wong, M. W.; Andres, J. L.; Replogle, E. S.; Gomperts, R.; Martin, R. L.; Fox, D. J.;
Binkley, J. S.; Defrees,
D. J.; Baker, J.; Stewart, J. P.; Head-Gordon, M.; Gonzalez, C.; Pople, J. A. Gaussian 94, revision D.4; Gaussian, Inc.: Pittsburgh,
PA, 1995.
15. Beigler-Kőming, F. W.; Bader, R. F. W.; Tang T. H. J. Comput. Chem., 3, 317, 1982. (b) AIMPAC: A suite of programs for the Theory
of Atoms in Molecules;R.F.W. Bader and coworkers (Eds.), McMaster University, Hamilton, Ont., Canada L8S 4M1. Contact bader@mcmail.cis.mcmaster.ca.
16. Graña, A.M.; Mosquera, R.A. J. Mol. Struct., 556, 69-76, 2000.
17. Popelier, P.L.A.; Aicken F.M.A. Can. J. Chem. 78: 415-426, 2000.
18. 1 Hartree = 627.5095 Kcal / mol
19. Graña, A.M.; Mosquera, R.A. J. Chem. Phys., 110, 1999.