Introducción
En la actualidad a los organismos vivos como las plantas y los animales no se les puede considerar como individuos o entidades biológicas independientes definidas mediante criterios anatómicos o fisiológicos, debido a que una gran diversidad de organismos simbiontes se encuentra habitando cada una de estas entidades biológicas para completar sus vías metabólicas y así, cumplir funciones fisiológicas. Por otra parte, los organismos simbiontes también constituyen un segundo modo de herencia genética que proporciona y complementa la variación genética del organismo hospedero, el cual finalmente es sometido a los mecanismos de la selección natural (Gilbert et al., 2012).
Entre los microorganismos que podemos encontrar como simbiontes se encuentran las bacterias, las arqueas, los virus y los organismos protistas que, en conjunto con el hospedero, forman el holobionte (Raina et al., 2018). El holobionte se considera, de manera general y conceptual, como un organismo eucarionte, planta o animal, que incluye a todos sus microorganismos simbióticos asociados que forman unidades anatómicas, fisiológicas, inmunológicas y evolutivas en la naturaleza (Simon et al., 2019a). El termino holobionte deriva del griego antiguo ὅλος (hólos) que significa «todos» y βιὅς, que significa “organismo”, “ser vivo” (Revista Ciencia y Desarrollo, 2018). El concepto de holobionte fue introducido por Lynn Margulis en 1991, sin embargo, se ha propuesto que dicho término fue usado desde 1943 por el biólogo teórico alemán Adolf Meyer‐Abich (Baedke et al., 2020), lo que sugiere que este concepto ha sido utilizado por la comunidad científica desde hace más de 50 años.
De la misma manera, el concepto de holobionte ha surgido en el marco teórico y experimental para estudiar las interacciones entre hospederos y sus comunidades microbianas, las cuales evolucionan y se desarrollan en todo tipo de ecosistemas. Actualmente, se han establecido varios modelos biológicos de estudio, en donde se han incluido a los humanos, las plantas, las esponjas y los insectos, para determinar el papel del holobionte en la biología, la ecología y la evolución del hospedero (Simon et al., 2019b; van de Guchte et al., 2018). Asimismo, se ha observado que en el proceso evolutivo del holobionte pueden ocurrir cambios en el genoma del hospedero como en cualquiera de los genomas microbianos asociados al mismo, y que depende de la cooperación entre los genomas dentro del holobionte, así como de la competencia con otros holobiontes para que se desarrollen en un nicho ecológico (Theis et al., 2016).
A partir de las evidencias científicas antes mencionadas, Richard Jefferson en 1994 propuso el término de hologenoma, mismo que en 2007 fue propuesto de forma independiente por Eugene Rosenberg y Llana Zilber-Rosenberg (Bordenstein & Theis, 2015). El concepto de hologenoma se refiere al contenido genético completo del hospedero, de sus organelos y de todo su microbioma, el cual puede ser transferido entre ellos y varía entre cada holobionte. En este sentido, el hologenoma es todo el sistema genético del holobionte, que aumenta o disminuye la salud física del hospedero. Además, dentro de las subunidades genómicas contenidas en el hologenoma constantemente surgen las mutaciones a sus propias tasas finitas, tal es el caso del genoma nuclear en el que la selección natural distingue entre variantes genéticas favorables y no favorables, determinando cuales de estas mutaciones servirán para mejorar la aptitud del holobionte. Por ejemplo, en el microbioma la selección favorece la propagación de microbios benéficos involucrados en la nutrición, la defensa y su reproducción (Douglas, 2015), mientras que los microorganismos patógenos son eliminados a través de la selección natural, aunque en ocasiones estos mismos desarrollan adaptaciones para mejorar su transmisión en la próxima generación del hospedero (LePage & Bordenstein, 2013; Ma et al., 2014). En resumen, el hospedero puede adquirir nuevos microorganismos provenientes del ambiente o ampliar la abundancia microbiana por medio de la transferencia genética horizontal entre organismos del microbioma del hospedero, lo cual genera un efecto directo sobre la variabilidad genética del hologenoma.
Finalmente, se ha establecido que existe una estrecha relación entre el desarrollo de enfermedades y el microbioma de los seres humanos. El microbioma de los humanos el cual se compone en su mayoría por bacterias, ha llamado la atención en los últimos años debido a que existen varios reportes científicos que han sugerido que el microbioma puede contribuir al riesgo de desarrollar cáncer (Gopalakrishnan et al., 2018). En esta revisión nos enfocaremos en dar un panorama general sobre los últimos estudios del hologenoma y su relación con el desarrollo del cáncer a través del microbioma. Además, se mencionarán las distintas técnicas de secuenciación de ácidos nucleicos que han sido utilizadas actualmente para tratar de entender al holobionte a través del estudio de la hologenómica para finalmente hacer especial énfasis en los análisis bioinformáticos los cuales han apoyado para entender la hologenómica y su relación con el cáncer para el desarrollo de nuevas herramientas diagnósticas y su aplicación en la práctica clínica.
1. Concepto de microbioma
Durante más de un siglo las enfermedades se han clasificado en infecciosas y no infecciosas, con base en los postulados de Robert Koch, con la finalidad de establecer una relación causal entre un agente infeccioso específico y su enfermedad particular. Este acercamiento permitió avanzar en la identificación y comprensión de distintas enfermedades y sus tratamientos, sin embargo, contribuyó a mantener un panorama incompleto de los factores involucrados en el inicio y desarrollo de una enfermedad (Pitlik & Koren, 2017). Durante esos años, el estudio del microbioma estaba restringido a las técnicas dependientes del cultivo celular, que modificaban la composición original del microbioma y limitaban el análisis únicamente a especies con características particulares que les permitían crecer en cultivo (Shanahan et al., 2020), excluyendo especies difíciles de cultivar que incluían bacterias intracelulares como Coxiella burnetiior o Tropheryma whipplei las cuales requieren células hospedadoras para poder sobrevivir y dividirse (Bonnet et al., 2020).
Durante la última década, el estudio del microbioma ha adquirido gran importancia y se le define como aquellos microorganismos que viven, habitan e interactúan entre sí en un mismo entorno. En este mismo concepto también se incluyen los genomas y distintos metabolitos que genera cada especie del microbioma, así como el ambiente donde se desarrollan y su interacción con el organismo hospedero. Entre las diversas comunidades de microorganismos que habitan el cuerpo humano, en su mayoría corresponde a una gran diversidad de bacterias, principalmente del filo Firmicutes, Actinobacteria y Bacteroidetes (The Human Microbiome Project Consortium, 2012), así como una gran diversidad de hongos, dentro de los que destacan los géneros Candida, Saccharomyces y Cladosporium (Underhill & Iliev, 2014). Además, se ha visto que la composición y distribución del microbioma es heterogénea en el humano (Schwabe & Jobin, 2013). Por otro lado, el estudio del microbioma se puede abordar desde diferentes perspectivas, y técnicas, entre ellas, se encuentran la secuenciación masiva en paralelo del DNA, la cual ha permitido estudiar el conjunto de los diferentes genomas que componen al microbioma (Lynch & Pedersen, 2016). El acercamiento, desde un punto de vista genómico, ha permitido realizar un análisis más amplio sobre la composición y distribución del microbioma en humanos, así como realizar el análisis de metadatos sobre el parentesco genético y el éxito reproductivo relativo dentro de las poblaciones en el microbioma, con lo cual se han podido estudiar e identificar diferentes condiciones extrínsecas que influyen sobre el microbioma y, a su vez, el efecto que tienen estos cambios sobre la salud humana. Parte de la gran importancia que ha adquirido el estudio del microbioma ha sido el hecho de que se han encontrado asociaciones entre los cambios en el microbioma y diversas enfermedades, lo cual ha cambiado la visión general que se tenía sobre la relación entre enfermedad, salud y microbioma.
Actualmente podríamos considerar a cualquier enfermedad como la interacción disfuncional entre los diferentes factores ambientales tales como la dieta, el estilo de vida, la composición del microbioma y la genética del hospedero (Apidianakis & Ferrandon, 2014). Esta definición se sustenta en la creciente evidencia que relaciona diversas patologías con las alteraciones en la composición, la distribución y la función del microbioma, dichas alteraciones son conocidas como disbiosis (AlHilli & Bae-Jump, 2020). A la fecha, se han descrito tres tipos: disbiosis por proliferación de patobiontes en donde se incluyen las bacterias de los géneros Klebsiella y Enterobacter (Kitamoto, s. f.), disbiosis por pérdida de microorganismos comensales que incluye diferentes especies de Lactobacillus (Brusselaers, s. f.) y disbiosis por pérdida de diversidad, que implica la pérdida de especies de Lactobacillus provocando disbiosis vaginal (van de Wijgert & Jespers, 2017). Estos tres tipos de disbiosis no son mutuamente excluyentes y puede haber situaciones en las que se presente más de un tipo a la vez (Levy et al., 2017). Asimismo, el estudio de los distintos tipos de disbiosis y su relación con el desarrollo de diversas enfermedades multifactoriales (J. Wang & Jia, 2016), ha sido impulsado en gran parte mediante el análisis de datos de microbioma incluso en enfermedades consideradas como no infecciosas, como las metabólicas, la diabetes y la obesidad (Fan & Pedersen, 2020); así como enfermedades neurológicas que incluyen el autismo y la depresión (Ezra-Nevo et al., 2020; Marx et al., 2020) e incluso el cáncer (Elinav et al., 2019; Vogtmann & Goedert, 2016) enfermedad donde el estudio de los factores que la promueven ha sido tema central en su investigación durante los últimos años y donde existe una estrecha relación de su desarrollo con la composición de microbioma.
2. Cáncer
El cáncer se ha definido como un conjunto de enfermedades heterogéneas caracterizadas por la presencia de alteraciones a nivel genético y epigenético que afectan los mecanismos celulares regulatorios y de homeostasis, tales como el crecimiento, la división y la diferenciación celular, entre otros (Ginsburg et al., 2020). Los factores que originan el cáncer han sido motivo de estudio durante varios años y se sabe que existen diferentes factores de riesgo involucrados en su desarrollo, tales como la edad (Rizzi et al., 2020), las infecciones virales (Golrokh Mofrad et al., 2020; Nelson & Benson, 2017), la exposición a radiación (Akbani et al., 2015), la exposición a contaminantes ambientales (Hwang et al., 2020), y la dieta (AlHilli & Bae-Jump, 2020; Lien & Vander Heiden, 2019), entre otros, siendo esta última un factor que ha adquirido gran importancia en los últimos años (Steck & Murphy, 2020), debido a que se ha observado que los cambios en la dieta pueden originar predisposición al desarrollo de cáncer, o por el contrario, ofrecer efectos protectores contra este padecimiento. Por tal motivo, el estudio sobre la influencia que tiene la dieta en el desarrollo de cáncer ha cobrado gran importancia actualmente.
3. Asociación de la alimentación y el desarrollo de cáncer
Como se mencionó anteriormente, a través de diversos estudios se ha comprobado la asociación entre la dieta y el desarrollo de cáncer, donde se ha observado que la dieta puede tener efectos benéficos y ayudar en la prevención del desarrollo o progresión de la enfermedad (Bail et al., 2016) así como potenciar el efecto de las terapias contra el cáncer (Kanarek et al., 2020). Sin embargo, también se ha establecido que la dieta puede tener un efecto adverso, convirtiéndose en un factor de riesgo para el desarrollo de esta enfermedad (Lien & Vander Heiden, 2019; Steck & Murphy, 2020). Se ha comprobado, que a nivel molecular, la dieta puede tener un efecto en la producción de especies reactivas de oxígeno (Sun et al., 2020; Tobore, 2020), variaciones en la disponibilidad de nutrientes (Lien & Vander Heiden, 2019), cambios en el metabolismo celular (Bose et al., 2020) y cambios importantes en el microbioma (AlHilli & Bae-Jump, 2020; Bisanz et al., 2019; Polo et al., 2020). El estudio del microbioma y su asociación con el desarrollo del cáncer ha sido de vital importancia ya que se ha comprobado la regulación de la homeostasis de manera importante (Baquero & Nombela, 2012). Por lo tanto, los cambios en la composición y distribución del microbioma pueden estar asociados al desarrollo de enfermedades multifactoriales como el cáncer.
Como se mencionó anteriormente, la disbiosis, es uno de los factores predisponentes a cáncer (Biragyn & Ferrucci, 2018; Sharma et al., 2020); se sabe que al menos un 20% de todos los casos de cáncer a nivel mundial están asociados a esta condición (Pevsner-Fischer et al., 2016). Además de los factores como la dieta, la actividad física, el estilo de vida y la genética del hospedero (Manor et al., 2020; Schmidt et al., 2018; J. Wang & Jia, 2016), la edad es otro de los factores importantes que provocan cambios en el microbioma (Biragyn & Ferrucci, 2018), los cuales tienen un gran impacto sobre el metabolismo, la respuesta inmune, así como en procesos inflamatorios, siendo estas últimas tres parte de las marcas distintivas del cáncer (Hanahan & Weinberg, 2011).
4. Relación del microbioma y el cáncer
Uno de los principales tipos de cáncer asociado al microbioma es el cáncer del tracto gastrointestinal. Por ejemplo, las infecciones bacterianas por la bacteria Helicobacter pylori, están fuertemente asociadas al desarrollo de cáncer gástrico (Malfertheiner et al., 2014; Noh et al., 2020). Se ha visto que esta bacteria patógena promueve estrés replicativo en las células de la mucosa gástrica, promoviendo el daño al DNA (Bauer et al., 2020), la inestabilidad genómica, y por consiguiente el desarrollo de cáncer gástrico. (Cassidy & Venkitaraman, 2012; Pikor et al., 2013). Sin embargo, este ejemplo representa una enfermedad asociada a una única bacteria patógena presente en el tracto gastrointestinal, pero existen otros ejemplos como el cáncer colorrectal (CCR) donde se han caracterizado, mediante el análisis del microbioma, cambios en la abundancia de distintas especies bacterianas presentes en tejido tumoral. El CCR presenta una disbiosis característica en la que se ha encontrado una abundancia mayor de microorganismos pro-carcinogénicos como Fusobacterium nucleatum y Escherichia coli, y una menor cantidad de bacterias consideradas “protectoras” como Roseburia y Clostridium (Janney et al., 2020). Como resultado, el microbioma se ha propuesto como un biomarcador para el CCR, siendo mayormente asociados los géneros Fusobacterium, Porphyromonas (Shindo et al., 2019), y Peptostreptococcus (Ternes et al., 2020). Dentro del género Fusobacterium, el efecto de F. nucleatum ha sido ampliamente estudiado a nivel molecular y se ha visto asociado a la proliferación celular, una de las marcas distintivas del cáncer, esta bacteria puede promover la proliferación de células cancerosas por distintas vías de señalización intracelular, por ejemplo, se ha comprobado que activa al factor nuclear kappa B (NFkB) induciendo la sobreexpresión del microRNA-21 (Yang et al., 2017) el cual suprime la expresión del gen RASA1 (RAS P21 Protein Activator 1), y en donde la disminución en sus niveles de expresión origina la activación de la vía de MAPK (RAS-mitogen-activated protein kinase) induciendo un aumento en la proliferación celular. Además de esta vía, también se ha comprobado que puede promover la proliferación celular a través de la vía de señalización de Wnt, como se verá más adelante a detalle (Rubinstein et al., 2013). Por tal motivo, en este ejemplo de CCR, se puede observar la influencia del microbioma a nivel molecular en el desarrollo de cáncer.
Otro de los ejemplos de cáncer, donde se sabe que la disbiosis promueve el desarrollo de otra de sus marcas distintivas, la inflamación, es el carcinoma oral de células escamosas (COCE). La inflamación crónica es una condición que se ha asociado a cáncer durante varios años (Hanahan & Weinberg, 2011), ya que promueve la liberación de factores de crecimiento que inducen la proliferación celular y también la liberación de enzimas de matriz extracelular que facilitan la invasión, la metástasis y la angiogénesis (Dias et al., 2021). En COCE, se ha observado en saliva que un aumento en el conteo de las bacterias Porphyromonas gingivalis, Fusobacterium nucleatum y Prevotella intermedia, mediante la técnica de checkerboard DNA-DNA hybridization (Mager et al., 2005) están asociados a periodontitis que es un tipo de inflamación en las encías (Gholizadeh et al., 2016). En este caso, se ha propuesto al microbioma como un posible biomarcador de diagnóstico ya que en las muestras de saliva se ha observado una gran abundancia de las bacterias Capnocytophaga gingivalis, Prevotella melaninogenica y Streptococcus mitis, por lo que podría ser un indicador diagnóstico de COCE (Mager et al., 2005).
Hasta ahora se ha comprobado la relación entre el microbioma y el desarrollo de algunas neoplasias, en general se sabe que existen diversos factores intrínsecos del hospedero como la edad y la genética, o cambios ambientales como la dieta y el estilo de vida que pueden originar variaciones en la distribución y diversidad del microbioma, y en algunos casos originar disbiosis (Figura 1A), una condición que puede promover inflamación o estrés oxidativo (Figura 1B), estando estas dos condiciones asociadas al desarrollo de cáncer como marcas distintivas (Figura 1C). La identificación de los cambios en la composición del microbioma se puede identificar de diversas maneras, inicialmente se pueden utilizar técnicas como el cultivo en agares sólidos que permiten la identificación y aislamiento de colonias específicas de bacterias u hongos, pero a su vez dificultan la identificación de microorganismos de difícil crecimiento en cultivo como bacterias intracelulares. Además de estas técnicas de cultivo, las técnicas para la identificación de microorganismos basadas en el análisis de ácidos nucleicos como PCR o checkerboard DNA-DNA hybridization, permiten identificar microorganismos sin la necesidad de ser cultivados, por ejemplo, bacterias de interés clínico para la detección de COCE como Capnocytophaga gingivalis, Prevotella melaninogenica y Streptococcus mitis (Mager et al., 2005). Adicionalmente, el desarrollo de técnicas de secuenciación masiva de DNA han hecho posible identificar diversas poblaciones de microorganismos presentes en el cuerpo humano y el análisis de su abundancia y distribución, por ejemplo, mediante el análisis de RNA ribosomal 16s (Takahashi et al., 2019)because the mortality rate has recently decreased in other developed countries. The impact of microbiota in carcinogenesis, especially in the digestive tract has been reported. This study aimed to clarify the relationship between oral cancer and oral microbiota in Japanese patients.\nMethods: DNA was extracted from salivary samples of 60 oral cancer patients and 80 non-cancer individuals as controls. We performed metagenomic analysis using 16S rRNA amplicon sequencing. Statistical analysis in this study was performed using R (version 3.5.0 o el análisis de metagenoma (Shotgun metagenomics) (S. Yu et al., 2021)three with moderately severe acute pancreatitis (MSAP, técnicas que permiten un acercamiento más amplio para el estudio de las diferentes comunidades de microorganismos presentes en una muestra y no solo un enfoque a una única especie o un género de microorganismo.
Figura 1
Influencia del microbioma en el desarrollo y progresión del cáncer
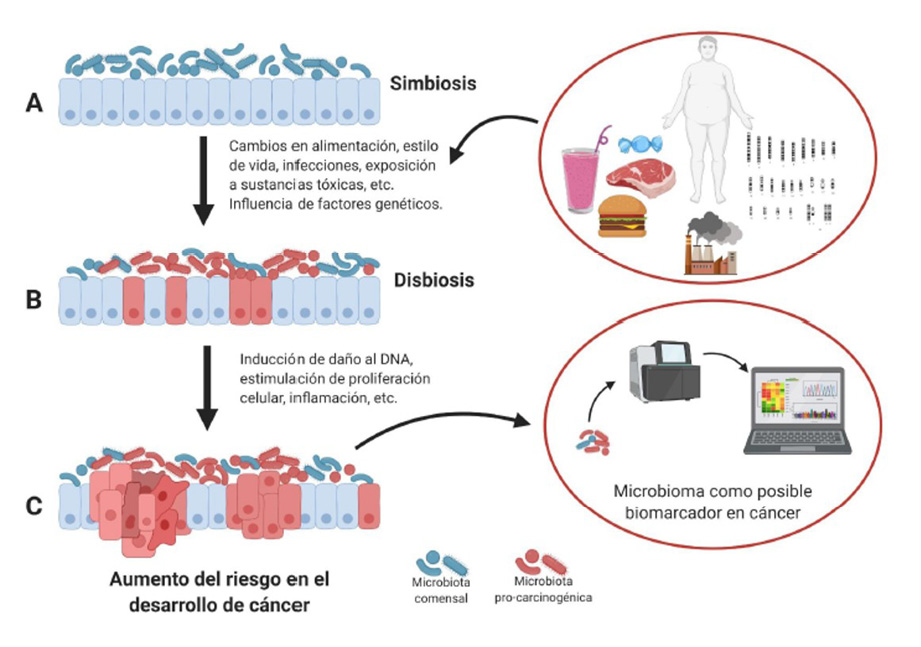
A) En un estado de simbiosis hay un equilibrio adecuado del microbioma comensal en el cuerpo humano, sin embargo, cambios en factores intrínsecos como influencia de factores genéticos, o extrínsecos como cambios en la dieta, estilo de vida, infecciones o exposición a contaminantes ambientales, entre otros, pueden inducir la pérdida de este equilibrio y favorecer un estado de disbiosis. B) En un estado de disbiosis hay una pérdida del equilibrio en el microbioma, se puede observar una o varias de las siguientes situaciones: proliferación de patobiontes, pérdida de microbioma comensal o pérdida de diversidad. Algunas bacterias patógenas pueden favorecer la aparición de marcas distintivas del cáncer, Helicobacter pylori puede inducir daño al DNA o, en el caso de la bacteria Fusobacterium nucleatum, un aumento en la proliferación. Otras bacterias como Porphyromonas gingivalis pueden inducir inflamación, estas tres bacterias están asociadas al desarrollo de cáncer gástrico, cáncer colorrectal y carcinoma oral de células escamosas, respectivamente. C) Se ha comprobado que la disbiosis puede inducir el desarrollo de cáncer mediante diferentes vías, por ende se ha propuesto al microbioma como un posible biomarcador para el diagnóstico de diferentes neoplasias entre las que destacan el cáncer colorrectal donde se puede ver una abundancia mayor de bacterias como F. nucleatum, también en cáncer gástrico donde se observa la presencia de H. pylori o tipos de cáncer como el COCE donde el conjunto de varias especies de bacterias (Capnocytophaga gingivalis, Prevotella melaninogenica y Streptococcus mitis) se pueden utilizar como un biomarcador de diagnóstico.
5. Microbioma y su relación con la regulación de las características distintivas del cáncer (Hallmarks)
Se ha propuesto la presencia de 8 rasgos distintivos del cáncer que describen las capacidades que adquieren las células tumorales en su evolución hacia el desarrollo de una neoplasia y que les permite sobrevivir, proliferar y propagarse. Estas características son: la auto señalización de proliferación sostenida, la insensibilidad ante inhibidores de crecimiento, la evasión de la apoptosis, la angiogénesis sostenida, la capacidad de invasión tisular, así como la generación de metástasis, la adquisición de potencial replicativo ilimitado, la reprogramación del metabolismo energético y la evasión de destrucción por el sistema inmune (Pevsner-Fischer et al., 2016). Recientemente, se ha encontrado evidencia que sugiere la participación del microbioma para promover la adquisición de estos rasgos distintivos del cáncer, como veremos a continuación (Schmidt et al., 2018).
Los procesos de proliferación y muerte celular están regulados por diversas señales que deben mantener un balance para conservar la arquitectura y función de los tejidos. En el caso de la mucosa intestinal se ha comprobado que diversas bacterias comúnmente encontradas en el tracto digestivo tienen un efecto en este balance, como Bacteroides fragilis y Fusobacterium nucleatum, que mediante la interacción con la proteína de adhesión celular E-Caderina, pueden inducir la proliferación de células epiteliales al activar la vía intracelular de señalización Wnt/Beta-Catenina y llevar a cabo la transcripción del proto-oncogen c-Myc (Gholizadeh et al., 2016; Yang et al., 2017). Asimismo, se demostró que las cepas de la bacteria E. coli portadoras de la isla pks (pks+ E. coli), comúnmente encontradas en tumores colorrectales, inducen a las células senescentes a secretar el factor de crecimiento de hepatocitos (HGF). La presencia de HGF lleva a la proliferación de células epiteliales mediante la expresión del microRNA-20a-5p que tiene como blanco a SENP1, que regula la SUMOilación de la proteína P53, uno de los principales reguladores del ciclo celular y la apoptosis (Mager et al., 2005). Esta relación entre el microbioma y la proteína de P53 evidencia un posible mecanismo para la adquisición de algunas de las características distintivas del cáncer, como son la proliferación celular sostenida y la evasión de señales de apoptosis.
Por consiguiente, al haber una mayor proliferación y menor apoptosis en tejidos neoplásicos hay un incremento general del número de células, por lo que requieren un mayor suministro de nutrientes. Por este motivo, la angiogénesis sostenida es esencial para el mantenimiento, el crecimiento y la propagación de células tumorales, siendo otra de las características distintivas del cáncer que puede ser inducida por el microbioma. La evidencia experimental ha demostrado que durante un proceso infeccioso las bacterias, hongos, virus y protozoarios pueden inducir la angiogénesis ya sea mediante la estimulación directa de moléculas del patógeno sobre el hospedero, como en el caso de los lipopolisacáridos propios de bacterias gramnegativas, que son reconocidos por el sistema inmune innato y estimulan la producción de citocinas proinflamatorias; o de manera indirecta en respuesta a un proceso inflamatorio o daño celular (Ding & Schloss, 2014). Por otra parte, en ratones criados en ausencia de microorganismos se observa un arresto en el desarrollo de redes capilares, que puede ser reactivado y corregido en tan solo 10 días después de la inoculación de microbioma de ratones sanos o cultivos de Bacteroides thetaiotaomicron (Limborg et al., 2018), demostrando que la interacción del microbioma con su hospedero no sólo influye, sino que es esencial para la angiogénesis durante el desarrollo del intestino y probablemente en procesos neoplásicos.
También se ha estudiado la interacción del microbioma con el sistema inmune en las mucosas. Se ha demostrado que la presencia de microorganismos, como son B. fragilis y varias especies del género Alistipes pueden inducir una respuesta inmune con presencia abundante de células T colaboradoras productoras de interleucina-17 (Th17), que se asocian a la activación de vías proinflamatorias como la de Stat3, mayor crecimiento tumoral y metástasis en cáncer colorrectal (Tierney et al., 2019). Asimismo, el microbioma puede interactuar también de manera directa con las células del sistema inmune, como es el caso de F. nucleatum, que interactúa mediante proteínas de adhesión Fap2 con los receptores de células T, inhibiendo su capacidad citotóxica y con esto su respuesta antitumoral (Plaza Oñate et al., 2019). Esta evidencia demuestra que el microbioma puede regular al sistema inmune ayudando a las células neoplásicas a evadir su destrucción, que es otra de las características distintivas del cáncer. Sin embargo, el microbioma no siempre tiene un efecto protumoral, los microorganismos específicos como los del género Bifidobacterium, pueden estimular las capacidades citotóxicas de las células T, promoviendo su actividad antitumoral (Forslund et al., 2015). De este modo, el microbioma puede regular al sistema inmune hacia una respuesta proinflamatoria o antiinflamatoria dependiendo su composición, lo que sugiere el uso de posibles tratamientos basados en la generación de una respuesta inmune antitumoral mediante la manipulación específica del microbioma.
Otra posible línea de tratamiento consiste en utilizar el microbioma para generar un ambiente antitumoral aprovechando la desregulación del metabolismo energético que es una de las características de las células neoplásicas. A diferencia de la mayoría de las células en el organismo que de manera general basan su metabolismo en la fosforilación oxidativa, las células tumorales exhiben un consumo acelerado de glucosa que oxidan parcialmente a lactato mediante glucólisis aerobia, lo que se conoce como efecto Warburg (Zolfo et al., 2017). Las células epiteliales en el colon, por otra parte, pueden basar su metabolismo hasta en un 70% en oxidación del butirato que se encuentre presente en el tracto digestivo. El butirato es producido por el microbioma a través de la fermentación de fibra dietaria, que se define como los componentes de las plantas que no pueden ser metabolizadas por las enzimas digestivas de los humanos. Los polisacáridos no amiláceos que contiene la fibra dietaria son procesados por el microbioma en el tracto digestivo, produciendo ácidos grasos de cadena corta como el propio butirato, que ha demostrado tener un efecto anti-tumoral (Truong et al., 2015). El efecto antitumoral del butirato no consiste únicamente en no servir a las células tumorales como fuente de carbono, sino que al acumularse funciona como inhibidor de las desacetilasas de histonas y promueve la expresión de genes proapoptóticos como FAS y reguladores del ciclo celular, como p21 y p27, lo que en conjunto limita la proliferación celular y la progresión tumoral(Fan & Pedersen, 2020). De esta manera, se evidencia una estrecha relación entre la dieta y el microbioma en la generación de un microambiente antitumoral basado en el metabolismo energético característico de las células tumorales.
La evidencia experimental aquí revisada demuestra que hay una relación entre la composición del microbioma y la regulación de los mecanismos moleculares de cada una de las características distintivas del cáncer. En el caso de la mucosa intestinal, se ha sugerido que esta relación es establecida por la interacción directa e indirecta de los microorganismos en el tracto digestivo con diversos tipos celulares del hospedero, como son las células epiteliales y las células T del sistema inmune, para producir un efecto protumoral o antitumoral, dependiendo de la composición del microbioma. Asimismo, la necesidad de microorganismos en el tracto digestivo para el correcto desarrollo del intestino es una de las evidencias más sólidas para demostrar que la evolución del hospedero y su microbioma ha ocurrido de forma conjunta y, por lo tanto, el cáncer y otros padecimientos podrán ser comprendidos en su totalidad únicamente en el contexto del holobionte.
6. Flujos de trabajo y técnicas utilizadas en el análisis bioinformático de hologenomas
Como se ha mencionado antes, se ha descubierto que algunas funciones biológicas asociadas al microbioma contribuyen con el desarrollo de enfermedades consideradas como no infecciosas, por lo que ahora debemos entender cómo se han hecho este tipo de acercamientos y asociaciones. Sabemos que el holobionte es una entidad biológica constituida por microorganismos simbiontes y patógenos, cuya identidad biológica así como sus interacciones moleculares pueden ser descritas mediante el análisis integral de las biomoléculas que los componen, lo que se conoce como estudios ómicos, a través de técnicas como la secuenciación masiva en paralelo de DNA y RNA (Limborg et al., 2018).
Dada la complejidad de la muestra analizada, ya que contiene un conjunto de biomoléculas (ya sea DNA, RNA, proteínas o metabolitos) que pertenecen a los organismos asociados en comunidad en el holobionte, se han diseñado diferentes flujos de trabajo bioinformáticos (del inglés bioinformatic pipelines) para determinar si existen relaciones biológicas (como intercambios de metabolitos) entre dichos microorganismos y si éstas se asocian a funciones específicas tanto del hospedero como de los demás organismos comprendidos en el holobionte. Actualmente, la evidencia científica señala la existencia de relaciones funcionales (como las funciones metabólicas que aportan nutrientes esenciales) entre el hospedero y los otros organismos a nivel de epigenoma, genoma, transcriptoma, proteoma y metaboloma, que pudieran estar asociadas al desarrollo de enfermedades como el cáncer gastrointestinal, así como otros padecimientos crónico-degenerativos (Apidianakis & Ferrandon, 2014), como la diabetes (Forslund et al., 2015). En consecuencia, se han desarrollado metodologías para el análisis de las asociaciones funcionales descritas para los holobiontes de estos padecimientos, así como de su aplicación clínica en el desarrollo de fármacos y biomarcadores (Limborg et al., 2018).
Las asociaciones funcionales que existen entre el hospedero y los otros organismos que componen el holobionte se determinan calculando coeficientes de correlación que permiten identificar los componentes moleculares con los que cada organismo contribuye al hologenoma, así como las vías de señalización en las que se involucran, para lo cual se utilizan genomas de referencia de los diferentes componentes del holobionte. Inicialmente, el análisis del hologenoma incluye el flujo de trabajo de alineamiento, mapeo e identificación de unidades taxonómicas mediante el reconocimiento de regiones conocidas como marcos abiertos de lectura que son únicos para cada componente del holobionte (Tierney et al., 2019). El resultado de este proceso permite determinar la abundancia conjunta de los grupos de genes que deriva en la identificación de las diferentes especies metagenómicas presentes en el holobionte, así como los niveles taxonómicos a los que éstas pertenecen, mediante el uso de herramientas como MSPminer, que a partir de perfiles genómicos, lleva a cabo el análisis de co-abundancia de genes y permite identificar los organismos presentes en la muestra, tanto los ya caracterizados como los organismos no descritos antes para esa condición, lo que ha permitido la identificación de biomarcadores para el diagnóstico de enfermedades gastrointestinales así como nuevas especies en el hologenoma que no habían sido descritas (Plaza Oñate et al., 2019).
Asimismo, existen herramientas como StrainPhlAn, MetaMLST (Zolfo et al., 2017) y MetaPanPhlAn (Truong et al., 2015) que permiten hacer el análisis por tipo de organismo presente en el holobionte, aumentando la precisión al establecer relaciones funcionales, como se ha visto en el estudio del microbioma asociado a estómago en población europea que permitió la identificación de un nuevo perfil molecular asociado a Helycobacter pylori, lo que es relevante en el estudio de los padecimientos gastrointestinales y el desarrollo de biomarcadores novedosos (Fan & Pedersen, 2020). Además, se ha propuesto introducir el uso de clasificadores supervisados basados en aprendizaje de máquina (del inglés machine learning), que consiste en usar los diferentes lenguajes de programación para enseñarle a las computadoras cómo analizar y aprender de los datos para poder realizar predicciones o inferencias. Esto se hace con diferentes estrategias, como el algoritmo de bosques aleatorios que emplea la herramienta AUC-RF (Calle et al., 2011) y otros métodos como el descrito por Thompson y colaboradores (Thompson et al., 2019), que mejora la identificación de los organismos del holobionte, permitiendo además el descubrimiento de nuevas características del hologenoma que pueden tener implicaciones clínicas dentro del holobionte, lo que a su vez ha permitido la identificación de nuevos biomarcadores de diagnóstico, como se ha visto en el estudio de los componentes microbiológicos presentes cuando existen lesiones de colon pre-neoplásicas, demostrando los beneficios de la aplicación de estos nuevos enfoques bioinformáticos al estudio del hologenoma (Baxter et al., 2016; Flemer et al., 2018).
En cuanto a la información que utilizan estos flujos de trabajo, ésta proviene principalmente del análisis de datos de secuenciación, para lo cual se utilizan diferentes estrategias dependiendo de las características del holobionte que se pretenda estudiar. Particularmente, en el caso del hologenoma, éste es analizado comúnmente con secuenciación por fuerza bruta (o shotgun en inglés) y estudios de asociación de genoma completo (GWAS, por sus siglas en inglés), de cuyo análisis se deriva la determinación de la identidad de los organismos presentes en el holobionte (Fatkhullina et al., 2018). Sin embargo, se ha demostrado que los estudios de mayor utilidad corresponden a aquellos que usan los meta-GWAS (MGWAS), ya que aportan información relevante acerca de los cambios fenotípicos del hospedero asociados a genes microbianos específicos. Para ello, se requiere un genoma de referencia y conocer previamente el microbioma específico con el que se interactúa, lo que representa una dificultad si éstos no se conocen previamente o no han sido secuenciados. Por ejemplo, si se trata de un organismo cuyo genoma ya ha sido secuenciado, pero que no se había identificado previamente como parte del holobionte, el estudio se puede auxiliar de otros componentes del holobionte, como el holo-transcriptoma (Nyholm et al., 2020; Qin et al., 2012)
Por otro lado, con respecto al holo-transcriptoma asociado al holobionte éste, a diferencia del hologenoma, se define como el conjunto total de transcritos presente en el holobionte, y su estudio se puede abordar con experimentos de secuenciación masiva en paralelo de RNA pareada (del inglés paired-end) utilizando librerías del gen ribosomal 16s, cuya secuencia es específica entre los diferentes organismos y ha demostrado ser útil en la identificación de los microorganismos presentes en el tracto biliar que se relacionan con la condición neoplásica de este tejido (Lee et al., 2020). La secuenciación de tipo pareada del gen 16s se lleva a cabo con plataformas como MiSeq de Illumina (Kozich et al., 2013), para lo cual es necesario filtrar las secuencias para identificar los genes 16s rRNA con paquetes como Mothur (Schloss et al., 2009), y alineando con paquetes especiales como SILVA 16S rRNA. La identificación de los organismos del holobionte se puede realizar con los clasificadores bayesianos, como el descrito por Wang y colaboradores (Q. Wang et al., 2007), para finalmente identificar las unidades taxonómicas operacionales y con ello a los diferentes organismos presentes en la muestra evaluada, como se ha descrito en el estudio de Baxter y colaboradores, donde identificaron las unidades taxonómicas operacionales asociadas a Pophyromonas assaccharolytica, así como algunas otras bacterias presentes en el tracto gastrointestinal y cuya presencia puede utilizarse como biomarcador diagnóstico en padecimientos como el cáncer colorectal (Baxter et al., 2016). Además, se ha mostrado que no todos los componentes metagenómicos identificados a partir de la comparación de los rRNA 16S contribuyen en composición significativamente al hologenoma, por lo que es necesario el desarrollo de nuevas herramientas y metodologías bioinformáticas que permitan discriminar las relaciones significativas presentes entre los componentes del holobionte (Douglas & Werren, 2016)
Asimismo, en la determinación de las asociaciones estadísticamente significativas entre los componentes del holobionte, es decir, si estos componentes interactúan entre ellos como un sistema biológico integral, se utilizan de manera rutinaria análisis estadísticos como la prueba PERMANOVA, que se basa en el análisis multivariado llevado a cabo mediante permutaciones para determinar la respuesta simultánea de una o varias variables en un modelo de estudio con varios factores (Anderson, 2017). Incluso, ésta y otras herramientas se encuentran incluidas en una variedad de paqueterías informáticas diseñadas para análisis ecológico que se pueden implementar en el análisis de muestras clínicas en el estudio del hologenoma asociado a patologías (J. Yu et al., 2017), como es el caso de VEGAN (Dixon, 2003), que ha servido en la identificación de biomarcadores del hologenoma para la detección de lesiones cancerosas en el colon en estados tempranos (detectable desde la etapa temprana II (J. Yu et al., 2017) a través del uso de PERMANOVA, y DAME (Ding & Schloss, 2014), que es una plataforma en línea que permite analizar archivos de secuenciación de metagenomas de manera interactiva, lo que sugiere un avance práctico en las técnicas de diagnóstico para este padecimiento, ya que lo hace accesible a usuarios que no son expertos en bioinformática (Baxter et al., 2016). Entonces, la implementación de estas herramientas bioinformáticas en flujos de trabajo de investigación clínica, cuya funcionalidad ya ha sido comprobada en análisis ecológicos, permite ampliar el conocimiento del hologenoma para establecer su utilidad en el desarrollo de nuevos biomarcadores para padecimientos crónico degenerativos como la diabetes mellitus (Forslund et al., 2015; Qin et al., 2012), en donde también se ha incluido el cáncer (Apidianakis & Ferrandon, 2014; Flemer et al., 2018; Lee et al., 2020; J. Yu et al., 2017), lo que lleva a un avance en la medicina personalizada y llevando el manejo clínico de los pacientes de una manera óptima.
En resumen, el avance en las tecnologías de secuenciación, así como el desarrollo de nuevas herramientas bioinformáticas han permitido avanzar en la caracterización de los holobiontes, esto incluye desde mejorar la precisión con la cual se identifican los organismos presentes y la información genómica que puede adquirirse de ellos, hasta la caracterización de nuevos organismos no identificados previamente al conjuntar el análisis del hologenoma con el estudio ómico de otros componentes moleculares, como el transcriptoma. . Un ejemplo, es la identificación de una nueva cepa de Helicobacter pylori en el tracto gastrointestinal en población europea descrito por Fan y colaboradores (Fan & Pedersen, 2020) (Figura 2). Además, la implementación de herramientas bioinformáticas ya probadas en otras áreas como la ecología pueden ampliar la capacidad analítica del estudio del hologenoma y permitir su aplicación al ámbito clínico, como es el caso de los biomarcadores de diagnóstico en cáncer colorectal identificados por Baxter y colaboradores(Baxter et al., 2016), lo que representa un avance en el descubrimiento de nuevos biomarcadores moleculares, y lo cual contribuye con el desarrollo de la medicina de precisión en padecimientos como el cáncer y otras enfermedades crónico-degenerativas como la diabetes.
Figura 2
Análisis holo-ómicos
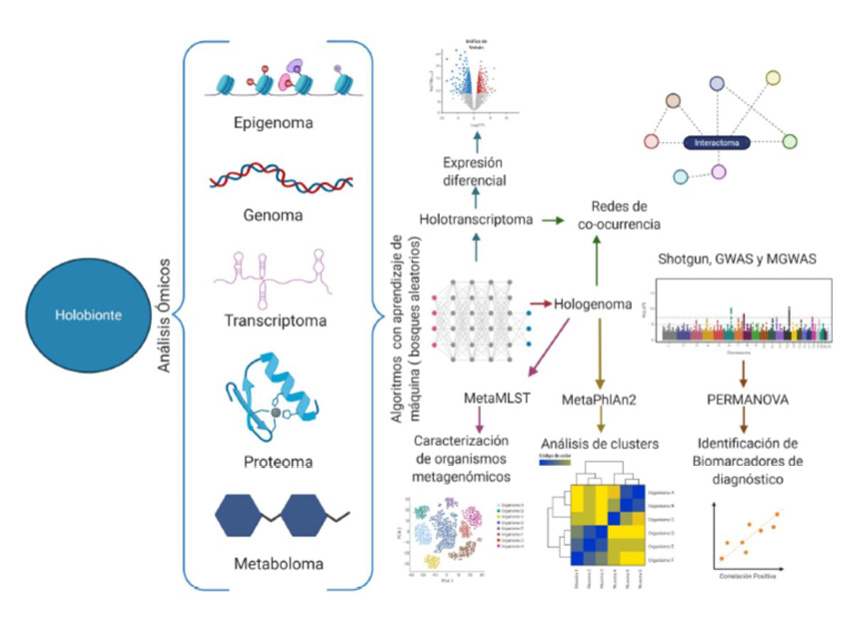
El estudio integrativo de los holobiontes permite establecer las asociaciones funcionales entre el hospedero y los otros organismos que componen este sistema biológico. Los flujos de trabajo bioinformáticos pueden mejorarse implementando el aprendizaje de máquina mediante el uso de bosques aleatorios para mejorar, por ejemplo, la identificación de los organismos presentes y sus características genómicas mediante diferentes tecnologías de secuenciación como shotgun, GWAS y MGWAS, para posteriormente identificar los grupos taxonómicos mediante análisis de clusters con herramientas como MetaPhlAn2 o mediante análisis multivariados como el utilizado con la herramienta MetaMLST, que se ha demostrado puede tener aplicaciones clínicas en el desarrollo de nuevos biomarcadores diagnósticos. Además, la integración del análisis de otros componentes moleculares del holobionte como el holotranscriptoma permiten la identificación de transcritos expresados en el holosistema mediante análisis de expresión diferencial, lo cual, en conjunto con la información del hologenoma, permite conocer las asociaciones funcionales entre los diferentes componentes del holobionte, lo que puede denominarse como el interactoma del holobionte.
7. Conclusiones
El holobionte es una entidad biológica en donde la asociación entre el hospedero y los simbiontes afectan la aptitud del holobionte dentro de su entorno, donde la variación o mutaciones en el hologenoma puede ser provocada por cambios en los genomas del hospedador o del microbioma y bajo estrés ambiental, la comunidad microbiana simbiótica puede cambiar rápidamente y es donde la visión del microbioma ha evolucionado bastante desde el desarrollo de las técnicas de secuenciación masiva, donde anteriormente el estudio del microbioma era realizado mediante la identificación de microorganismos con técnicas dependientes de cultivo, restringiendo este estudio únicamente a microorganismos con capacidad de crecimiento en cultivo. Al poder identificar y estudiar la composición y distribución del microbioma en el cuerpo humano mediante técnicas de secuenciación masiva en paralelo de ácidos nucleicos y la consecuente introducción del término “microbioma” se empezó a ver la compleja asociación del ser humano y su microbioma, por lo tanto, ahora se ve al ser humano como un holobionte el cual depende, para mantener su estado de homeostasis, de una adecuada distribución y diversidad en el microbioma que lo compone.
Es importante resaltar que, aunque algunos de los efectos del microbioma se deben a interacciones directas de los microorganismos con las células del intestino, una parte de los efectos se deben a la estimulación de una respuesta inflamatoria por parte del hospedero. En respuesta al daño en la mucosa intestinal, el Receptor del Factor de Crecimiento Epidérmico (EGFR) activa cascadas de señalización de algunas subfamilias de cinasas de proteínas activadas por mitógenos (MAPK), como ERK1/2, que estimula la proliferación celular y bloquea la apoptosis, lo que promueve la formación de tumores colorrectales. Una parte considerable de la investigación actual en prevención y tratamiento de cáncer colorrectal se centra en comprender los diversos factores que participan en activación de esta respuesta inflamatoria. Se ha demostrado, por ejemplo, que una dieta abundante en colesterol y grasas estimula una mayor producción de ácidos biliares, que son metabolizados por el microbioma en el intestino en ácidos biliares secundarios, como el ácido desoxicólico, que estimula la activación de EGFR y la proliferación celular. Algunas bacterias del género Clostridium, por otra parte, metabolizan los ácidos biliares en ácido ursodesoxicólico, que ha demostrado tener un papel protector contra el cáncer de colon y su administración a pacientes ha tenido resultados alentadores al prevenir la reaparición de tumores colorrectales.
Por otro lado, el metabolito producido por el microbioma que más extensamente ha sido estudiado por su efecto antitumoral es el butirato. Se ha demostrado que el butirato inhibe la desacetilasa de histona HDAC3, lo que lleva a la inactivación de las vías de Akt1 y Erk1/2, inhibiendo la proliferación y migración celular. Se descubrió de manera más reciente, que el butirato inhibe la transcripción del microRNA MiR-92a, lo que aumenta la expresión de genes supresores de tumores como la fosfatasa PTEN, que antagoniza la vía de PI3K, disminuyendo la proliferación celular y promoviendo la apoptosis. Asimismo, se vio que el butirato aumenta la transcripción de MiR-203, lo que disminuye la proliferación de la ubiquitina ligasa “Hakai”, disminuyendo la transición epitelio-mesénquima y la metástasis de células neoplásicas. La comprensión de estas nuevas vías de regulación conlleva a pensar en tratamientos no solo basados en la regulación de la alimentación o manipulación de la composición del microbioma, sino en el suministro directo de inhibidores, como el ácido ursodexicólico o incluso terapias génicas dirigidas. El uso de diversos anti-mirs, por ejemplo, para disminuir los niveles de MiR-92a ha mostrado resultados prometedores en modelos animales y podría dirigir el futuro de los tratamientos para cáncer de colon en los próximos años.
Entonces, los flujos de trabajo y técnicas bioinformáticas en el análisis del hologenoma, el desarrollo de nuevas herramientas tecnológicas de secuenciación, como la secuenciación de nueva generación, que actualmente permite el análisis de células individuales (del inglés single cell), permitirá la identificación individual con mayor precisión respecto a la resolución actual que proveen las técnicas de bulto (del inglés bulk) para el análisis de muestras complejas. No obstante, algunas limitaciones como el tamaño celular y las interacciones físicas de los microorganismos presentes en el holobionte pueden significar un obstáculo en la preparación de las librerías individuales, por lo que el desarrollo de nuevas metodologías de separación celular de muestras es uno de los retos actuales en el estudio de los holobiontes. No obstante, en las últimas dos décadas el análisis del hologenoma y del holotranscriptoma ha podido perfeccionarse, como se ha discutido en este texto, con el desarrollo de nuevas herramientas bioinformáticas capaces de identificar un mayor número de organismos presentes en los holobiontes, los cuales se incluyen en las bases de datos y que finalmente enriquecen con la adición de nuevas especies a este tipo de bases de acceso público. Actualmente, en la era de la secuenciación masiva, una de las premisas de esta área de estudio es el enriquecimiento de las bases de datos ya conocidas, ya que éstas son la fuente de referencias para la identificación de los organismos en el holobionte, aunque es preciso mencionar que uno de los mayores retos actuales para los investigadores y desarrolladores es el perfeccionamiento de herramientas bioinformáticas con una mayos sensibiliad y especificidad de sus métodos de análisis, que son los parámetros que permiten establecer la confiabilidad de los nuevos descubrimientos. En este sentido, uno de los aportes al perfeccionamiento de las herramientas bioinformáticas y los flujos de trabajo, ha sido la incorporación de algoritmos que incluyen el aprendizaje de máquina, dado que elimina el error humano y ha permitido la identificación de nuevos organismos que componen los holobiontes, como se ve descrito en el trabajo de Baxter y colaboradores donde se identificaron nuevos biomarcadores en cáncer colorectal, y lo cual permite el establecimiento de asociaciones funcionales de relevancia clínica en padecimientos como en el caso de cáncer de colon, y lo cual podría tener utilidad en el desarrollo de nuevos blancos terapéuticos así como la identificación de nuevos biomarcadores moleculares. Sin embargo, actualmente existe un número limitado de paqueterías específicas para el análisis de datos de secuenciación de hologenoma y holotranscriptoma con información clínica, y aunado a ello, existen pocos trabajos en los que se describa la incorporación de algoritmos con aprendizaje de máquina, por lo que una perspectiva para el desarrollo de flujos de trabajo bioinformático sería desarrollar nuevas plataformas o paqueterías amigables para usuarios no expertos en programación que permitan hacer análisis confiables de datos hologenómicos utilizando metadatos con información clínica para establecer asociaciones de utilidad clínica dentro del holobionte. Por ejemplo, las herramientas en línea que provee la plataforma Galaxy para análisis metagenómicos, o el caso de la aplicación web DAME. En ambos casos, ninguna de las plataformas es específica para el uso de datos clínicos, sin embargo, los flujos de trabajo pueden ser adaptados para obtener resultados clínicos relevantes y confiables en la identificación de organismos presentes en una muestra clínica, así como la identificación de nuevos biomarcadores o blancos terapéuticos. La recopilación específica de la información antes presentada también tiene como finalidad invitar al lector interesado en el análisis ómico del holobionte a que explore las nuevas posibilidades que le proporcionan las herramientas bioinformáticas descritas en este trabajo y que las identifique como nuevas oportunidades de análisis en el estudio de holobiontes con relevancia clínica. Asimismo, la incorporación rutinaria de algoritmos que utilicen el aprendizaje de máquina representa una alternativa analítica novedosa que permitirá la identificación de asociaciones funcionales estadísticamente significativas que no han sido descritas, lo cual contribuye con la descripción molecular y funcional de los sistemas holobiontes y proporcionará información de utilidad en el desarrollo de nuevas herramientas diagnósticas y su aplicación en la práctica clínica, lo cual también es un área de oportunidad para los desarrolladores de paqueterías y herramientas bioinformáticas, ya que como hemos mencionado antes, no existen paqueterías específicas, ni plataformas, para el análisis de muestras de holobiontes de relevancia clínica que sean amigables con usuarios no expertos en programación.
Finalmente, es indispensable seguir estudiando la relación entre el ser humano y su microbioma, así como su influencia con el desarrollo de cáncer con ayuda de nuevas plataformas de secuenciación y nuevas paqueterías bioinformáticas, para tener una visión global sobre el desarrollo de esta enfermedad y en un futuro utilizar al microbioma como una herramienta clínica a nuestro favor, ya sea como un posible biomarcador o un blanco terapéutico para el manejo clínico del paciente oncológico.
Agradecimientos
Agradecemos el apoyo de la Dra. María del Rocío Arellano Llamas y la M. en C. Clementina Castro Hernández por la lectura crítica y revisión de este artículo. Se agradece al Instituto Nacional de Cancerología (INCan) por su apoyo. Las figuras se crearon con BioRender.com.
Fuentes de Financiamiento
Laura Contreras-Espinosa es estudiante de maestría en el “Programa de Posgrado en Ciencias Biológicas, UNAM” y recibió una beca del Consejo Nacional de Ciencia y Tecnología (CONACYT) con CVU 1003211. Rogelio Montiel-Manríquez es estudiante de doctorado en el “Programa de Doctorado en Ciencias Biomédicas, UNAM con CVU: 581151.