Introducción
La historia de las enfermedades infecciosas representa un problema constante que afecta a millones de personas alrededor del mundo. Desde tiempos antiguos la humanidad ha enfrentado diferentes emergencias sanitarias que han resultado en pandemias afectando a decenas de países. En décadas recientes las de mayor impacto han sido de origen oriental. En 2002, en Guangdong el SARS-CoV (Síndrome respiratorio severo agudo), causó aproximadamente 700 decesos y más de 8000 personas contagiadas presentando una letalidad de 9.5%; en 2012, el Síndrome Respiratorio del Oriente Medio, causado por el MERS-CoV fue reportado en 27 países, con 2206 personas infectadas y 787 decesos, y una letalidad de 35.67% (Guarner, 2020; Hui et al., 2018). Actualmente, tenemos un virus agente causal de una enfermedad contagiosa que ha resultado en una pandemia mundial, para la cual, no tenemos ni medicamentos ni vacunas disponibles. Esta enfermedad es conocida como el nuevo síndrome respiratorio agudo severo, y es causado por el virus SARS-CoV-2 (COVID-19), que según datos de la Organización Mundial de la Salud (OMS), tuvo su brote en la localidad de Wuhan en la República Popular de China en diciembre de 2019 (WHO, 2020).
En respuesta, la comunidad científica ha propuesto una serie de diferentes estrategias que generen información que contribuya al combate del SARS-CoV-2. Al respecto, se han realizado secuenciaciones del genoma de aproximadamente 8275 virus asilados de pacientes de diversos países del mundo. Gracias a esta herramienta, se puede acceder a una mejor información para el avance en el desarrollo de medicamentos y vacunas. En este sentido, la aplicación de la secuenciación genómica nos acerca a conocer y descifrar la información del genoma de los agentes causales de diversas enfermedades infecciosas. Adicionalmente, la dilucidación de dicho genoma, permite tener al alcance información de los diversos rasgos biológicos que componen a estos virus, con el fin de comprender y predecir su comportamiento.
1. Antecedentes del COVID-19
Los coronavirus de SARS-CoV-2 (COVID-19), son virus de RNA monocatenario positivo (+ssRNA) de 27 a 32 kb, que son capaces de replicarse en ausencia de DNA. El RNA genómico posee una cap en la región 5´terminal, mientras que en la región 3´ presenta diversas poliadenilaciones. Estos virus poseen una estructura esférica o pleomórfica de 80 a 120 nm de diámetro y una nucleocápside helicoidal formada por glicoproteínas organizadas por proyecciones de trímeros de la glicoproteína S (Spike) y dímeros de proteínas HE (Hemaglutininas-Esterasas), responsables del reconocimiento a los receptores ACE2 de las células del hospedero (Masters, 2006). En su genoma existen de 6-11 marcos de lectura abierta (ORF´s), que codifican para proteínas no estructurales y accesorias (Figura 1) (Cui, Li, & Shi, 2019).
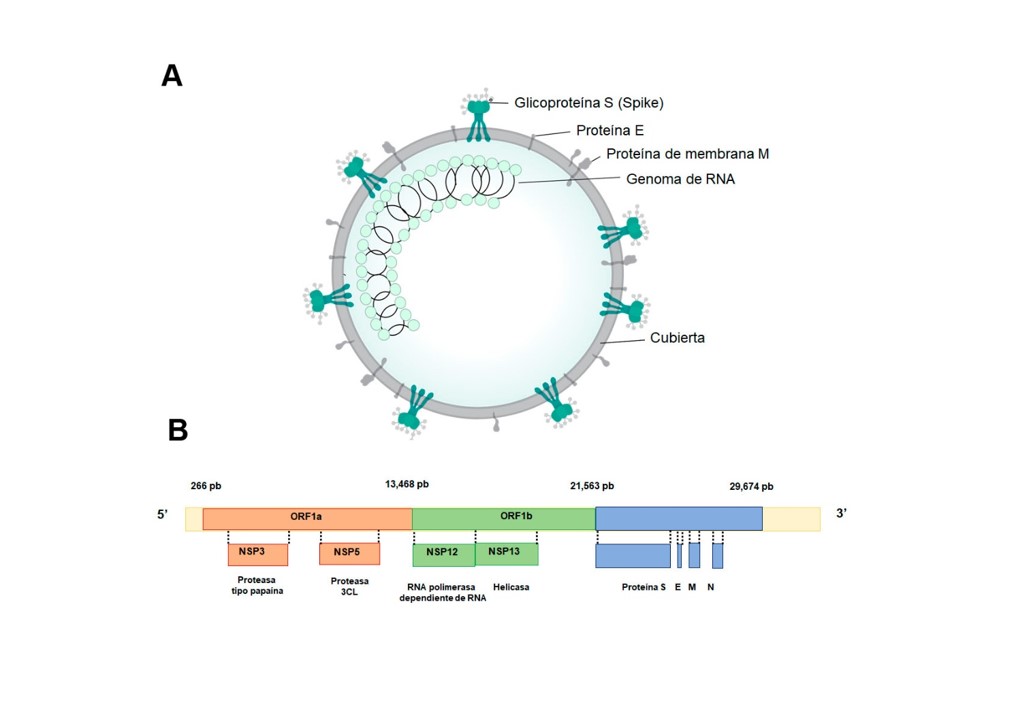 |
Proteínas estructurales y organización del genoma del COVID-19. A) sección transversal con las principales proteínas estructurales. Modificada al español de By SPQR10Binte altaf - Own work, CC BY-SA 4.0, https://commons.wikimedia.org/w/index.php?curid=88349537. B) Genoma de COVID-19 organizado en ORF1a, ORF1b y ORF de proteínas estructurales S (Spike), E (cubierta), M (membrane) y N (nucleocápside). |
Los coronavirus pertenecen a la familia Coronaviridae y al orden de los Nidovirales (Huang et al., 2020), de los cuales, los géneros alfa y beta, infectan a los humanos causando enfermedades respiratorias y gastroenteritis en animales, mientras que los delta y gamma, infectan a las aves, originando múltiples afecciones (Cui et al., 2019).
Estos virus se replican en las células del tracto respiratorio, aunque al momento existen reportes de la capacidad invasiva en diversos tipos de células epiteliales (Huang et al., 2020; Hui et al., 2018). En el ser humano, el desarrollo de la infección consta de un periodo de incubación promedio de aproximadamente 6 días dentro de un rango de 2 a 11. El crecimiento y la expansión de la infección es de tipo exponencial, y se estima que cada persona infectada es capaz de contagiar aproximadamente de 2 a 3 individuos dentro del periodo de incubación. No obstante, es importante considerar que, a 30°C, los coronavirus son capaces de seguir siendo infecciosos en diferentes superficies hasta por 9 días. Adicionalmente, el COVID-19 es considerado como un virus con una alta tasa de contagio y una tasa de mortalidad de 1.4 a 3.7% (Huang et al., 2020).
2. Impacto a nivel mundial y datos en México
En diciembre de 2019, el departamento de salud en Wuhan, República Popular de China, reportó diferentes casos de neumonía de agente etiológico desconocido. En respuesta, el Centro Chino para el Control y Prevención de Enfermedades (CDC-China), en conjunto con autoridades sanitarias, realizaron diferentes pruebas moleculares para determinar el agente causal de dichos padecimientos. Basados en la secuenciación genómica del virus se encontró que se trataba de un coronavirus perteneciente al género de los Beta-Coronavirus (Liu, Chen, & Chen, 2019), cuyas micrografías de microscopía electrónica revelaron estructuras virales de 60 a 140 nm con las espículas de superficie (Glicoproteínas S), distintivas a los virus de la familia Coronaviridae. Consecuentemente, un nuevo coronavirus denominado SARS-CoV-2 (COVID-19) fue anunciado en China, como el agente etiológico de la neumonía atípica presentada en los pacientes reportados. Desde entonces y hasta mayo del 2020, se han reportado aproximadamente 4,097,894 casos confirmados de COVID-19, y 311,588 decesos en 185 países del mundo, con una letalidad de 6.78%. En el continente americano particularmente, se registran 2,049,566 de infectados y más de 123,091 muerte, con una tasa de letalidad de 6.01%. En México, el primer caso de COVID-19 se registró el 27 de febrero en pacientes que provenían de Italia con destino a Ciudad de México y Sinaloa. Hasta el 28 de mayo, según datos de la Secretaría de Salud en México existen 78,023 casos registrados, con 8,597 decesos y 15,592 casos activos.
3. Diagnóstico
El objetivo primordial de las pruebas diagnósticas radica en la detección del origen de las neumonías para poder contribuir al control de la enfermedad y su diseminación. Para esto, la prueba más utilizada considerada como el estándar de oro, es la reacción en cadena de la polimerasa de transcriptasa reversa (RT-PCR). La prueba consiste en la detección de la secuencia de un gen de la envoltura del virus, no obstante, la sensibilidad y especificidad de esta prueba aún no se conoce con certeza, pero el algoritmo diagnóstico, requiere la detección de secuencias del gen de la nucleoproteína o de la RNApol viral. Cabe resaltar que las variaciones obtenidas en los resultados, también dependen del sitio de donde es tomada la muestra (bronquial, aspirado nasal, aspirado faríngeo, heces o sangre). Un factor poco considerado para las pruebas diagnósticas, es el relacionado con las variaciones genéticas intrínsecas al virus, ya que se han detectado varios subtipos.
A la fecha, no se conoce al paciente cero, sin embargo, se piensa que este virus podría provenir de animales silvestres (Forster, Forster, Renfrew, & Forster, 2020), el origen zoonótico del virus es una incógnita y sigue siendo investigado, ya que el COVID-19 presenta porcentajes significativos de identidad genómica con el coronavirus de diversas especies de murciélagos (Zhou et al., 2020).
Existen pocos estudios acerca de la asociación que existe entre la infección por COVID-19 y la presencia de otros patógenos, por lo que resulta de suma importancia el desarrollo de nuevas estrategias moleculares y de secuenciación que coadyuven en el conocimiento de los factores genéticos asociados, a identificar el potencial origen de la enfermedad y su interacción con otros patógenos, entre otros aspectos.
4. Metagenómica como herramienta para la detección de patógenos
La secuenciación de nueva generación (NGS, por sus siglas en inglés), permite la identificación de miles a billones de fragmentos de DNA/RNA. Una de las aplicaciones de la NGS en la microbiología clínica es la metagenómica NGS (mNGS), que representa un abordaje para la detección de patógenos a partir de muestras clínicas de pacientes, animales, muestras ambientales, etc. (Figura 2) (Gu, Miller, & Chiu, 2019). La metagenómica es el análisis genómico de secuencias de una población de microorganismos, que hace posible conocer el perfil de las comunidades microbianas en el medio ambiente y su interacción con el cuerpo humano (Flygare et al., 2016). Un análisis metagenómico tiene el potencial para detectar patógenos nuevos y raros y descubrir el desbalance en los microbiomas en las infecciones humanas, así como también, permite realizar inferencias sobre resistencia a antibióticos, virulencia, evolución y los posibles mecanismos de transmisión (Figura 3) (Miller, Montoya, Gardy, Patrick, & Tang, 2013).
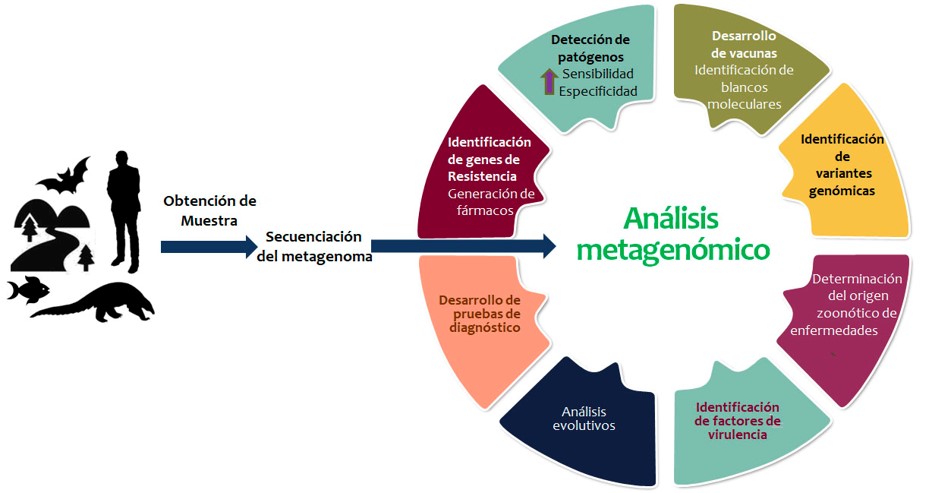 |
Aplicaciones potenciales del análisis metagenómico a partir de distintos tipos de muestras (ambientales, animales, humanas). |
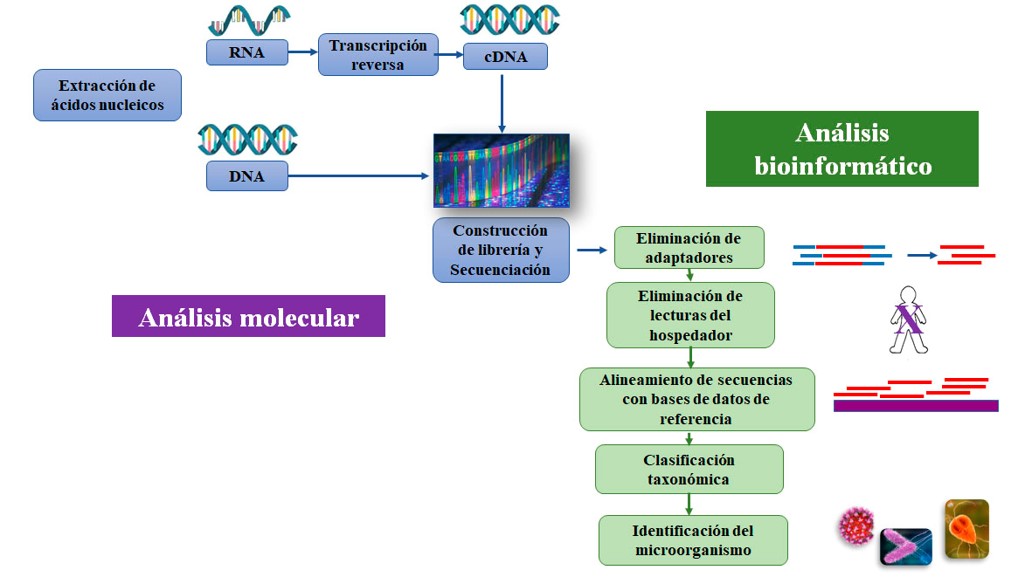 |
Diagrama de flujo para el análisis metagenómico de muestras. El desarrollo de un análisis metagenómico consta básicamente de dos etapas, una que consiste en la obtención del material genético (DNA y RNA), donde se incluye un análisis molecular de las muestras; y una segunda etapa que constituye el análisis bioinformático. |
En comparación con los métodos de diagnóstico tradicionales, la mNGS tiene la ventaja de identificar patógenos esperados, no esperados e incluso nuevos microorganismos (Chiu, 2013), permitir la identificación de nuevas cepas, predecir la resistencia a fármacos e incluso proveer datos cuantitativos de la concentración de microorganismos en la muestra a partir del conteo de las lecturas secuenciadas, lo que es muy útil en muestras polimicrobiales (Gu et al., 2019). Dentro de las desventajas inherentes a esta técnica está la presencia de lecturas de DNA del propio paciente (hasta >99%) lo que limita su sensibilidad.
5. Estructura metodológica para el análisis metagenómico
de las muestras, esto incluye el aislamiento del DNA y/o RNA. Estas muestras deben estar libres de contaminantes (DNA/RNA del hospedero como plantas, animal o ser humano), ya que la menor cantidad de éstos en las muestras disminuyen el rendimiento de la extracción (Thomas et al 2012). El siguiente paso a considerar es la generación de secuencias de DNA a gran escala; este paso es fundamental y depende de la estrategia de secuenciación de nueva generación (Illumina, PacBio, Nanopore) que se haya seleccionado. El tercer paso del análisis metagenómico es el ensamblado de las secuencias, la cual puede ser basada en genomas de referencias o de novo (Escobar-Zepeda et al., 2018; Thomas et al., 2012). Actualmente, existen softwares disponibles que permiten el ensamblado basado en genomas de referencia, este tipo de ensamblado funciona bien si existen secuencias cercanas y requiere un consumo de tiempo relativamente sencillo. Por el contrario, el ensamblado de novo, demanda recursos computacionales poderosos ya que se procesan cantidades significativas de información sin tener un templado con el cual comparar. Una vez realizado el ensamblado del metagenoma, se procede a la anotación del mismo. Como primer paso, implica hacer uso de plataformas de anotación como MG-RAST, CAMERA o IMG/M que permiten identificar las proteínas codificadas en el metagenoma (predicción de características); como segundo paso, se asignan funciones a los genes y se determinan los vecinos taxonómicos (anotación funcional). El análisis estadístico y el almacenamiento de los datos son los pasos finales que se deben seguir. Debido a que los datos metagenómicos pueden contener un número mayor de especies o funciones de genes que el número de muestras tomadas, se deben evaluar pruebas de hipótesis múltiples. Y en cuanto al almacenamiento, esto involucra que la información se almacene y sea compartida con la comunidad científica para lograr un mejor nivel de organización y colaboración de los datos metagenómicos generados y centralizados en bases de datos (Escobar-Zepeda et al., 2018; Thomas et al., 2012).
Por otro lado, se han realizado algunos abordajes metagenómicos para la identificación de coronavirus, lo que ha llevado a contar con pruebas de diagnóstico confiables y relativamente rápidas (Li et al., 2020). Un estudio molecular sobre murciélagos en México, permitió la identificación de 13 coronavirus distintos, nueve de ellos del tipo alfa-coronavirus y cuatro del tipo beta-coronavirus, además de 12 nuevos (Anthony et al., 2013). Este estudio, basado en la identificación de genes específicos para coronavirus mediante PCR, mostró que una especie/género de murciélagos puede contener múltiples Coronavirus, uno de los beta-Coronavirus es 96.5% idéntico a nivel de aminoácidos con el beta-Coronavirus asociado con la enfermedad humana del síndrome respiratorio del Oriente Medio (MERS) y se encontró en el murciélago Nyctinomops laticaudatus. Este tipo de estudios refuerza la importancia de una constante vigilancia en las familias de murciélagos que fungen como reservorios de Coronavirus que afectan gravemente la salud humana.
Un estudio genómico sobre coronavirus reveló que el COVID-19 tiene un 79% de homología a nivel de nucleótidos con el virus SARS-CoV, así como también con dos coronavirus tipo SARS aislados de murciélagos herradura (Rhinolophus sinicus) localizados en China, con los que comparte un 89% de homología en nucleótidos; y con un tercer coronavirus del murciélago herradura intermediario (R. affinis) con quien muestra un 96% de la misma homología (Ceraolo & Giorgi, 2020). La identificación de Coronavirus mediante metagenómica no se restringe a murciélagos sino también a otros animales como la gallina de Guinea (Ducatez & Guerin, 2015), pangolines de Malasia (Lam et al., 2020; Liu et al., 2019), civetas de palmera Himalayas y perros mapache (Guan et al., 2003).
Para el diagnóstico de COVID-19, se han desarrollado métodos basados en la secuenciación metagenómica con MinION para identificar rápidamente (8 horas) el SARS-Cov-2, además de evaluar el microbioma en muestras nasofaríngeas de pacientes con COVID-19 (Moore et al., 2020).
Las aplicaciones de la metagenómica para el estudio de los coronavirus han tenido impacto en el diseño de vacunas basado en antígenos identificados mediante análisis metagenómicos. Recientemente, se ha reportado el diseño de una vacuna recombinante quimérica basada en epítopos antigénicos de las proteínas estructurales S, M y E del SARS-Cov-2 (Rahman et al., 2020). Este tipo de desarrollos vacunales se han basado en los estudios estructurales de proteínas del SARS-Cov, como la glicoproteína S (Spike). El conocimiento de este tipo de proteínas permite no solamente comprender la biología del coronavirus, sino proponer nuevos candidatos para tratamientos de prevención (Johnson, Graham, & Menachery, 2018). El uso de la metagenómica para el desarrollo de vacunas ha desvelado la evolución viral y su dinámica para lograr la identificación de proteínas como candidatos vacunales (Luciani, Bull, & Lloyd, 2012). El conocimiento estructural de las proteínas del COVID-19 a partir de su secuenciación ha permitido elucidar las mutaciones en el dominio de unión del receptor humano al SARS-Cov-2, ACE2 (Angiotensin-converting enzyme 2) (Andersen, Rambaut, Lipkin, Holmes, & Garry, 2020). Algunos blancos terapéuticos que se han señalado para el COVID-19 se encuentra la glicoproteína S (Spike) que se une al receptor humano ACE2, el cual facilita la fusión viral y su entrada a la célula, de tal forma que si se bloquea esta interacción se provee una estrategia para el potencial desarrollo de antivirales (Ullah et al., 2020).
Una ventaja de la metagenómica en el estudio de las infecciones virales es la capacidad de medir la frecuencia de las variantes virales en una población. Mediante estos estudios se han podido identificar hasta tres variantes virales de influenza AH1N1 en un paciente inmunosuprimido (Luciani et al., 2012). En murciélagos, se han identificado algunas variantes virales del coronavirus causante del MERS, algunas de las cuales son causantes de infecciones altamente persistentes (Banerjee et al., 2020). La metagenómica ha logrado elucidar la capacidad evolutiva de los virus de RNA, como los coronavirus, al ofrecer un panorama único sobre la rápida dinámica de adaptación que pueden presentar los virus en un hospedero infectado, y con ello, la oportunidad de investigar en un periodo corto de tiempo el papel de la inmunidad adaptativa del sistema inmune durante esta dinámica evolutiva (Luciani et al., 2012). En general, la identificación de las variantes virales permitirá el desarrollo de fármacos o vacunas dirigidas específicamente contra variantes propias de una población.
Los fármacos antivirales empleados contra el SARS-CoV-2, se han desarrollado a partir del conocimiento de las secuencias y estructuras de las proteínas involucradas en el ciclo de vida del virus. Se ha propuesto que los mejores blancos terapéuticos son la proteasa tipo quimiotripsina, la proteasa tipo papaína, la helicasa y la RNA polimerasa dependiente de RNA, así como la glicoproteína estructural S. Los análisis se las secuencias genómicas de los coronavirus causantes del SARS y el MERS han identificado los sitios catalíticos y los sitios de unión de fármacos clave en las enzimas mencionadas, proponiendo así tratamientos contra el COVID-19 (Ullah et al., 2020).
6. Conclusiones
La presencia de nuevos patógenos que afectan la salud humana, como el COVID-19, resalta la importancia de contar con alternativas metodológicas que permitan conocer la información contenida en su genoma, con la finalidad de detectarlos, de conocer su interacción con otros patógenos, su potencial origen zoonótico, hasta la identificación de variables genómicas, la presencia de factores de virulencia y genes de resistencia a fármacos. La metagenómica se presenta como una opción viable para alcanzar este nivel de conocimiento, ya que sus potenciales aplicaciones generarán datos e información valiosa, especialmente cuando ocurren brotes de enfermedades que requieren una pronta respuesta y el uso de metodologías poderosas para la obtención de información. Adicionalmente, la bioinformática se enfrenta al reto de desarrollar mejores plataformas que contengan herramientas sofisticadas, incluyendo bases de datos para ensamble y anotación de metagenomas, plataformas de secuenciación más poderosas, y la preparación de recursos humanos confinados a la ejecución de análisis que contribuyan a la obtención resultados y aproximaciones más exactas.