Introduction
The COVID-19 pandemic in the first half of 2020 (Zhu et al., 2020) has put incredible pressure on all sectors of society, especially on health-care teams (physicians, nurses, and clinical analysts) as well as on scientific research teams. Suddenly, a worldwide effort was focused on developing methods for diagnosis, preventive vaccines, and molecules for treatment for those patients with more severe symptoms. Modern Medicinal Chemistry techniques found a critical challenge, and different strategies were used, including the repositioning of existing drugs, high-throughput screening, and computational techniques. In this latter sense, a large number of molecular docking papers began to appear in the literature. These studies aimed to find candidates that could be used in the clinic or were the start of accelerated programs towards a cure (ul Qamar et al., 2020; Wu et al., 2020). Several groups have published their results in open-science platforms or preprints even before peer review, providing very needed information for the synthesis and bioevaluation of the most promising compounds (Zastrow, 2020).
Several conclusions emerged from these studies. First, if some structures could be found, additional optimization of the computational hit would be needed. Secondly, molecular docking is a useful computational tool, but it should be complemented with additional studies like molecular dynamics simulations to provide additional support to the findings of structure-based virtual screening (SBVS). One of the most important conclusions is that there are molecules already used in the clinic or from natural products that could have therapeutic use against COVID-19, then it is not like all efforts must start from scratch.
From homology studies, it was revealed the high grade of similarity between four relevant enzymes for the cycle of the SARS-CoV-2 with those of the causative agent of SARS: 3 -chymotrypsin-like protease or main protease (Mpro, also known as Nsp 5), the papain-like protease (PLpro, Nsp3), RNA dependent RNA polymerase (RdRp, Nsp 12) and helicase (Nsp 13) (Wu et al., 2020). Hence, the initial step of this investigation was to locate which molecules have been evaluated against the SARS causative agent, whose outbreak appeared in 2002 and the virus causing MERS, even though the latter has a lower degree of homology with SARS-CoV-2. The search on the literature showed that various compounds were developed, targeting the four targets mentioned above. Our attention was drawn to certain compounds based on derivatives of chromone 1 as helicase inhibitors (Lee et al., 2009), cinnanserin 2 as an inhibitor of Mpro (Chen et al., 2005), and certain derivatives of naphthalene 3 as inhibitors of PLpro (Báez-Santos et al., 2015). Recently the antiviral activity of certain derivatives was reported 4-quinolone 4 against MERS- CoV (Yoon et al., 2019), so this would represent another interesting nucleus, although its activity against SARS- CoV or SARS-CoV-2 or its potential mechanism of action has not been demonstrated. A molecular design based on the common structural features of these compounds could render a scaffold for the generation of new and interesting candidates.
In our group, we have carried out several studies on compounds in which their structures resemble those of compounds 1-4. Therefore, inspired by these structures, we decided to explore if some derivatives of anthranilic acid 6, quinazolines 7, and phenylpropanoids 8, could have a theoretical affinity for any of the four targets of SARS CoV and SARS CoV-2. For the choice of substituents, it was decided for this initial exploration that all the molecules conserve the catechol part of the helicase inhibitor 1 and the cinnamic acid portion of cinnanserin 2. Compounds bearing this molecular template have been studied for their potent anti-inflammatory and antioxidant activities (Lopes-Lutz et al., 2008; Ruan et al., 2017). This attribute could give the studied compounds additional desirable properties, as it has been reported that dysfunctional immune responses lead to the progression of the most severe cases. Thus, immunomodulatory compounds could complete the therapeutic approaches (Tay et al., 2020). In this paper, we present the in silico evaluation through molecular docking of the compounds 5-8 (Figure 1). A molecular dynamics study was carried out for the most promising compounds to obtain more information that contributes to the development of new antivirals.
 |
Compounds with demonstrated in vitro activity on some of the SARS and MERS enzymes (1-4) and compounds for initial screening (5-8). |
1. Methodology
1.1 Molecular docking.
A previously reported methodology was used (M. Loza-Mejía et al., 2018; M. A. Loza-Mejía & Salazar, 2015). All molecules 1-9 and the active metabolite of remdesivir, GS-441524, which was included as reference ligand for the RdRp, were constructed using the program Spartan’10 (Wavefunction Inc, n.d.) and their geometry was optimized using MMFF // ab initio 6-31G*. Then, the optimized structures were exported to Molegro Virtual Docker (CLC Bio, n.d.; Thomsen & Christensen, 2006) and to YASARA Dynamics (Krieger & Vriend, 2014, 2015; Yasara Dynamics, n.d.) to carry out the molecular docking studies using the search and scoring algorithms integrated into each program (it is worth mentioning that YASARA uses the search algorithms of Autodock Vina (Trott & Olson, 2009) and scoring based on the binding energy) using 50 runs in each case; two different programs were used to find consensus poses (Houston & Walkinshaw, 2013). The structures of the target enzymes were downloaded from the Protein Data Bank (Berman et al., 2000), and before docking procedures, all water and co-crystallization molecules were removed. The selection of search sites was based on the information available at the time of initiating this work. In the case of Mpro (PDB code: 6LU7), the searching site was situated on subsite S1, which is delimited by residues 140–145 and 163–166 (Li et al., 2016). For PLpro (PDB code: 6W9C), it has been described that most of the enzymes’ competitive inhibitors bind on the subsites named S2 and S4 (Báez-Santos et al., 2014). Based on this information, the search site was located in the cavity formed between the residues 162-166 and the residues 271-273. In RdRp (PDB code: 6M71) the searching site was ubicated on the cavity formed by residues 618, 680, 760 and 761, which was based on the comparative analysis of the binding mode of sofosbuvir to the active site of the hepatitis C virus polymerase Ns5b, which was the base of the postulated binding mode of remdesivir to RdRp (Gao et al., 2020). Finally, the helicase search site (PDB code: 6JYT) was located on the ATP hydrolysis site delimited by residues 288-289, 374-375, 404, and 567 (Jia et al., 2019). In the case of Molegro Virtual Docker, the search site was delimited by a sphere of 12 Å in diameter and a grid of 0.3 Å, while for YASARA, it was a cuboid of 12 Å per side. Poses with the lowest score (MolDock Score for Molegro) or best binding energy (YASARA) were used for the subsequent analysis. First, the lowest poses for each combination ligand-enzyme in both programs were aligned using the MUSTANG algorithm and their RMSD calculated in YASARA Structure. If an RMSD value > 2.5 Å was recorded, we concluded that both programs generated a consensus pose.
1.2 Molecular dynamics
Molecular Dynamics (MD) simulations were performed to provide additional support to the molecular docking results. Simulations were carried out in YASARA Dynamics v.18.4.24 (Krieger & Vriend, 2015; Yasara Dynamics, n.d.) using the AMBER14 force field (Duan et al., 2003). The initial structures for the MD simulation were obtained from the docking complexes of compounds 7 -8 with the four enzymes analyzed in this work. The docking complexes of the compounds 1-3 in each of their target enzymes were also included in the MD studies to have a point of comparison. Although the affinity of these compounds against helicase, Mpro, or PLpro of SARS-CoV-2 has not been experimentally demonstrated, we consider that the degree similarity of the enzymes and those of SARS-CoV (above 88%) is enough to consider them as an initial reference. Each of the eleven complexes was positioned into a water box with a size of 100 Å × 100 Å × 100 Å, with periodic boundary conditions. The temperature was set at 298 K and water density to 0.997 g/cm3. Sodium (Na+) and chlorine (Cl−) ions were included, and pH was set to 7.4 to provide conditions that simulate a physiological solution (NaCl 0.9%). Particle Mesh Ewald (PME) algorithm was applied with a cut-off radius of 8 Å. The simulation snapshots were recorded at intervals of 100 ps with a timestep of 2.5 fs until a total simulation time of 30 ns was reached. It was considered that the complex reached equilibrium if RMSD variations were smaller than 2 Å for at least 20 ns. If the equilibrium was not reached for at least this period, the simulation time was increased. The MD trajectories were analyzed with a series of macros included as part of YASARA software, which included RMSD variations along time and ligand binding energy using MM-PBSA calculations and the snapshots of the last 20 ns were chosen for the calculation. This procedure has been previously reported (M. Loza-Mejía et al., 2018).
1.3 Prediction of ADME/Tox properties
For the prediction of the in silico ADME/Tox profile, the ADMETSar platform ( http://lmmd.ecust.edu.cn/admetsar2/ ) (Yang et al., 2018) was used, introducing the SMILES code of the four projected molecules as the base structure of new ligands for the studied enzymes. Mutagenic potential by AMES, inhibition of the hERG channel, passage through the blood-brain barrier, and absorption by the gastrointestinal tract. Additionally, the platform offers an evaluation called “in domain,” which is based on the prediction of six physicochemical properties related to oral absorption, including molecular weight, hydrogen bridge forming atoms (acceptors and donors), number of rings, logP and number of heavy atoms. Based on these properties, the platform determines whether or not these parameters are met.
2. Results
Table 1 shows the results obtained through molecular docking studies in both programs. The Rerank score values calculated with Molegro Virtual Docker (the lower the score value, the higher theoretical affinity), and the Ligand Binding Energy values (the more positive values, the higher the theoretical affinity) are shown. It can be appreciated that compounds 5-8 generally have higher theoretical affinity than reference ligands, regardless of the program used. Notably, our attention was drawn to ligands 7 and 8, which showed higher affinity in the four enzymes in comparison to the other two compounds, so the analysis was continued only for these two compounds. More conformational flexibility is required to be able to bind more than one enzyme, which would be expected in multi-target compounds (Talevi, 2015).
Table 1
Results of molecular docking studies
| Rerank score | Ligand (Kcal/mol) |
Binding | Energy | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | Mpro | PLpro | RdRp | Helicase | Mpro | PLpro | RdRp | Helicase | |
| 5 | -95.3 | -46.4 | -78.3 | -27.4 | 8.28 | 6.64 | 6.73 | 7.45 | |
| 6 | -121.6 | - 61.6 | -90.7 | -71.2 | 8.75 | 6.85 | 7.70 | 4.49 | |
| 7 | -122.8 | - 66. 5 | -93.4 | -94.6 | 8.26 | 6.72 | 7.84 | 5.92 | |
| 8 | -127.8 | - 81.0 | -111.4 | -86.0 | 7.91 | 6.97 | 7.94 | 6.57 | |
| Reference 1 | -116.1 | - 56.8 | -78.2 | -63.9 | 6.38 | 6.59 | 6.49 | 2.66 | |
1 Reference refers to ligand 1-3, whose in vitro activity against that enzyme is demonstrated. For Nsp12 (RdRp), the active metabolite of remdesevir was used as a reference.
In general, both programs predict similar poses for compound 7 with RMSD values near or below 2 Å, as shown in Table 2, so these poses can be used under the scheme of consensus docking (Houston & Walkinshaw, 2013; Torres et al., 2019) except for the poses predicted to PLpro. Higher differences were found in general for the complexes predicted for compound 8, which is predictable as the latter compound has high flexibility. When the RMSD difference is greater than 2 Å, it is impossible to decide which pose is convenient for further analysis. Despite this last fact, the analysis of the individual docking poses that both programs predict similar interaction patterns, as seen in Figures 2 and 3, with a more detailed analysis in the next paragraphs. Because of this fact, we decided to use the poses predicted by the YASARA program with the AutodockVina algorithm.
Table 2
RMSD values (Å) calculated for the poses predicted by the two docking programs
| Enzyme | Compound 7 | Compound 8 |
|---|---|---|
| Mpro | 2.31 | 2.62 |
| PLpro | 5.56 | 7.60 |
| RdRp | 1.33 | 11.1 |
| Helicase | 0.91 | 4.42 |
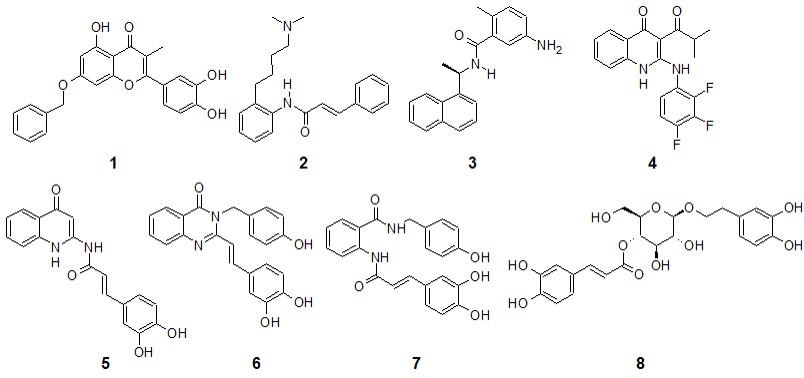
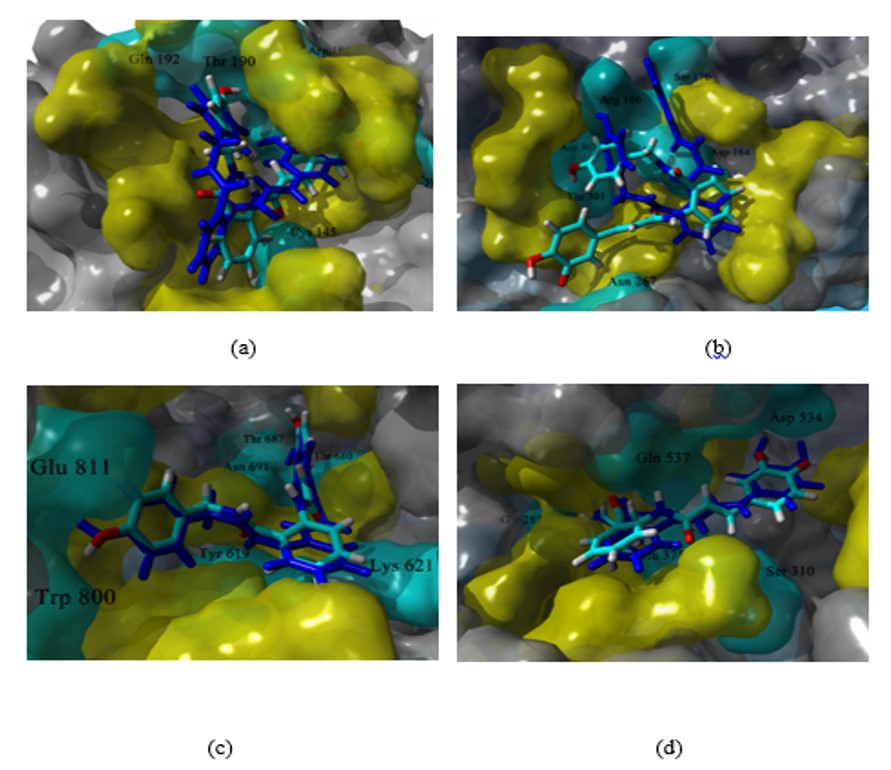
In the case of the poses predicted for Mpro (Figures 2a and 3a), it is interesting that both compounds adopt a V-shaped conformation within the cavity, which was also a similar observation found for some of the most promising candidates after a virtual screening analysis and MD studies carried out a molecular database of 606 million compounds (Fischer et al., 2020). Both compounds have a similar mode of interaction, as shown in forming hydrogen bonds with residues Arg188, Thr 190, and Gln 192. For poses predicted in the enzyme PLpro, there were no consensus poses. However, if it was possible to identify specific regions in common, it was again seen that compounds 7 and 8 adopt a V-shaped conformation (Figures 2b and 3b) with vertices in Ser 170, Asp 164 and Asp 267, except in the pose predicted by Molegro Virtual Docker algorithm, where compound 8 adopted a more linear conformation. On the other hand, in RdRp study both compounds again adopt a V-shaped conformation, the ends of the “V” is located, one of them between residues Trp 800 and Glu 811, while the other end in the cavity delimited by residues Thr 680, Thr 687 and Asn 691, as seen in Figures 2c and 3c. Finally, in the study of molecular docking on helicase, it was found that both compounds also adopt a V-shaped conformation, but the arrangement is different; for compound 7 the vertices of V are located at residues Gly 282, Glu 375 and Asp 534, while in compound 8 they are located between residues Glu 375, Ser 535 and Ala 313. From this analysis, it follows that compounds that can adopt a V-shaped conformation could interact more efficiently with the active sites of the four enzymes analyzed in this work.
 |
Poses predicted for compound 7 in the active site of MPro (a), PLpro (b), RdRp (c), and helicase (d). In CPK colors, the pose predicted in YASARA and blue the pose predicted by Molegro. In yellow and cyan, the residues relevant for the ligand-enzyme binding, in cyan and labeled, those that form a hydrogen bond. |
 |
Predicted poses for compound 8 at the active site of MPro (a), PLpro (b), RdRp (c), and helicase (d). In CPK colors, the pose predicted in YASARA and in blue the pose predicted by Molegro. In yellow and cyan, the residues relevant for the ligand-enzyme binding, in cyan and labeled, those that form a hydrogen bond. |
Figure 4 shows the variations of RMSD along time for the complexes analyzed during the MD studies. All complexes had relatively low variations from the initial docking pose (global RMSD after the first nanosecond of simulation < 2 Å), except by helicase complexes. Although these complexes reached equilibrium after 7 ns of simulation, they showed a higher degree of variation than the rest of the analyzed complexes, suggesting higher flexibility of helicase. The ligand-binding energy (LBE) was calculated from data of the snapshots of the last 20 ns of the simulation. This last data is shown in Table 3 for the eight complexes and the reference ligands 1-3. The algorithm implemented in YASARA has that ligands with more positive values of LBE have higher affinity. Thus, compound 7 has higher theoretical affinity against proteases Mpro and PLpro, than the reference ligands 1 and 2, while compound 8 against helicase, although the reference compound 3 has a higher affinity.
Table 3
Ligand Binding Energy values (Kcal/mol) calculated during the MD simulation studies1
| Compound | 7 | 8 | Reference |
|---|---|---|---|
| Mpro | 25.21 | -86.40 | 6.29 |
| PLpro | 59.32 | 0.39 | 48.50 |
| Helicase | -25.03 | 24.40 | 89.78 |
| RdRp | -14.50 | -123.00 | -11.28 |
1 More positive value indicate higher affinity.
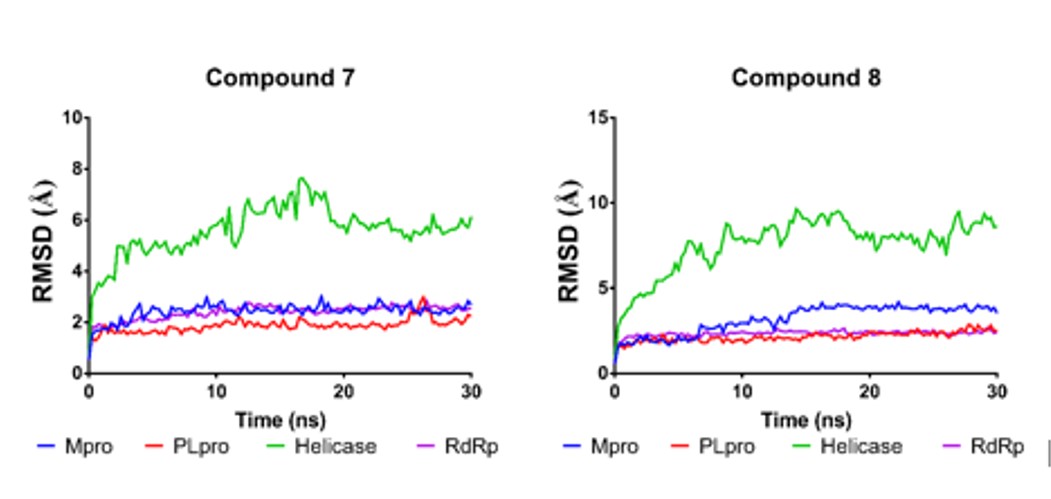 |
RMSD variations along simulation time for complexes of compound 7 and compound 8 with the four viral proteins analyzed. |
It is relevant to identify the changes in the interaction patterns before and after the MD simulations as it is valuable information for understanding the ligand-protein interaction since sometimes bindings modes are poorly represented on a “static” experiment such as docking. It is also essential to analyze the changes in the interaction pattern during the simulation time to identify the most stable interactions between the enzyme and the potential inhibitors. Figures 5 and 6 show the hydrogen bonding patterns in the poses generated from the molecular docking studies (with both algorithms implemented in Molegro and YASARA/Autodock Vina) and the structure after reaching the equilibrium in the MD simulation. As expected, some interactions were lost, and new ones were formed, particularly in the case of compound 8, but essentially both ligands remained within the enzyme cavity. Some conclusions were made from this analysis: a) The most stable interactions of compound 7 with the residues of the four enzymes, where those that involved the amide groups in the anthranilic acid core, as these interactions were maintained along all the simulation time; b) the higher flexibility of compound 8 could account for the differences between the interaction patterns obtained from docking studies and MD simulations and c) the V-shaped conformation was maintained despite these changes in the interaction pattern.
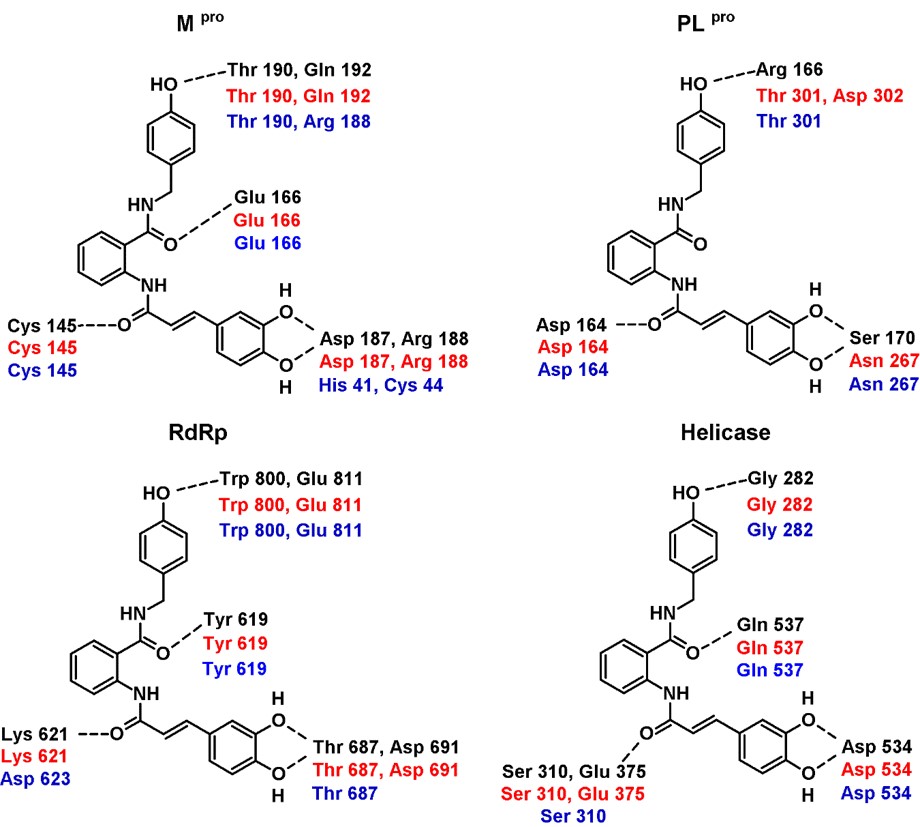 /> /> |
Hydrogen bonding interaction patterns for compound 7 in complex with the four targets analyzed in this study. In black are the residues that interacted in the best pose generated with Molegro Virtual Docker, in red those generated with YASARA/Autodock Vina, and in blue the interaction of the equilibrium complexes after the MD simulations. |
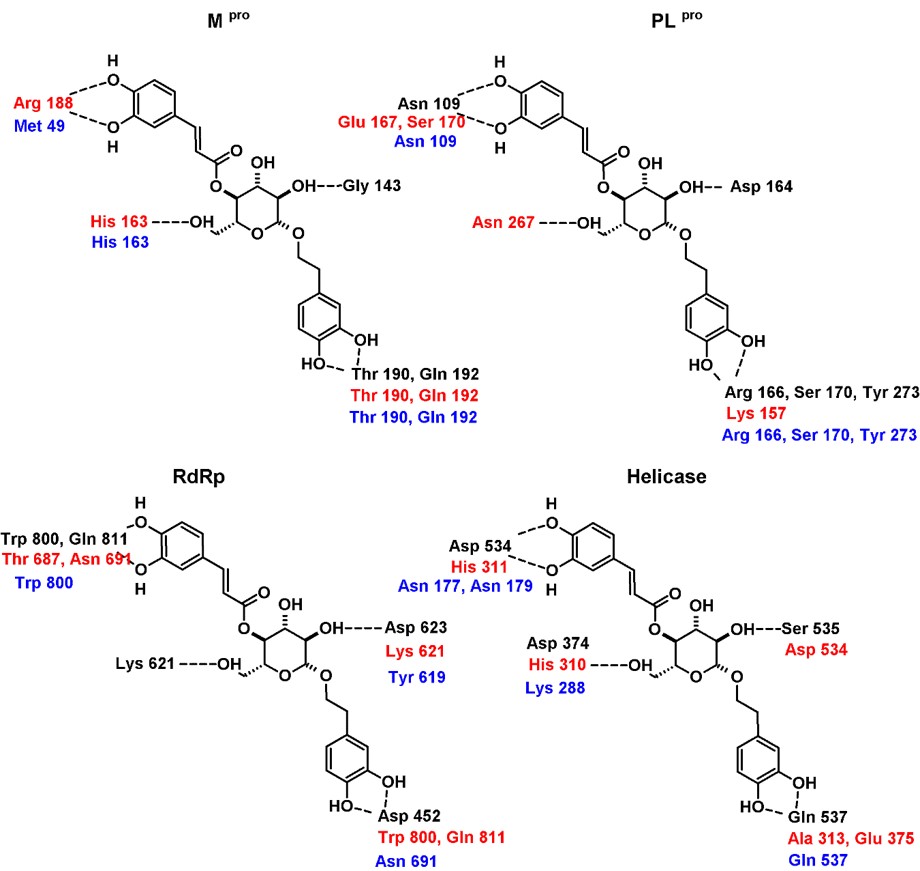 /> /> |
Hydrogen bonding interaction patterns for compound 8 in complex with the four targets analyzed in this study. In black are the residues that interacted in the best pose generated with Molegro Virtual Docker, in red those generated with YASARA/Autodock Vina, and in blue the interaction of the equilibrium complexes after the MD simulations. |
Something that should not be lost of sight of is that the pharmacokinetic-toxicological profile (ADME/Tox) could be a limitation for the development of new compounds. Table 4 shows the results of the predicted ADME/Tox profile of compounds 7 and 8. The evaluation given by ADMETSar suggests for compound 7, an acceptable pharmacokinetic profile, (absorption in the gastrointestinal tract, crosses the blood-brain barrier. promiscuity towards CYP450). However, the platform predicts problems hepatotoxicity, probably attributable to the presence of the α,β-unsaturated system, although that the presence of these groups can be considered as a “ warhead ” in some irreversible antiviral compounds. On the other hand, compound 8 would have absorption problems in the gastrointestinal tract, which would affect its oral absorption, although the rest of the ADME / Tox predicted parameters are acceptable.
Table 4
Prediction of ADME / Tox properties for compounds 7 and 8
| Compound | 7 | 8 |
|---|---|---|
| Gastrointestinal tract absorption | (+) | (-) |
| Blood-brain barrier passage | (+) | (+) |
| Glycoprotein-P substrate/inhibitor | (-) | (-) |
| CYP450 enzyme-substrate | CYP3A4 | CYP3A4 |
| Promiscuous CYP450 inhibitor | (-) | (-) |
| Ames test (mutagenicity) | (-) | (-) |
| Inhibitory channel hERG | (-) | (-) |
| Hepatotoxicity | (+) | (-) |
In conclusion, the analysis of the molecular docking and molecular dynamics calculations carried out on this small database shows that hydroxylated compounds with certain conformational flexibility that allows them to adopt a V-shaped conformation could be starting points for new antiviral development projects. The analysis of the interaction pattern suggests that the hydroxyl groups present in these molecules are needed for their interaction with some critical residues through hydrogen bonding. MM-PBSA ligand binding energy (LBE) calculations suggest that compound 7 could have good affinity against viral proteases and compound 8 against helicase. In addition to their potential antiviral activity, they have a favorable in silico ADME/Tox profile and could have significant anti-inflammatory and immunoregulatory activities based on their structural features. Thus, they could prevent virus proliferation and modulate some of the uncontrolled immune response, which has been recently recognized as a factor related to the most severe cases (Tang et al., 2020; Tay et al., 2020). An essential point of the efforts that have arisen from the crisis generated by COVID-19 is that they should not be interrupted. Much of the research-oriented towards the development of the cure, was born in basic science or is the continuation of projects that began with the study of other viruses. A Damocles sword is tingling above us: this is the third time that we have had an epidemic (now a pandemic) event due to coronaviruses in the 21st century (SARS, MERS, COVID), so efforts to find drugs that allow the treatment of the patients most affected by this or future coronaviruses is imperative.
Acknowledgments
No acknowledgments for this research project.
Funding
No funding was used for this research project.
Declaration of conflict of interest
The authors declare no conflict of interest with the publication of this manuscript.
Declaration of data availability
Data can be obtained from the correspondence authors.